




La miopatía necrosante inmunomediada (MNIM) es un subtipo de miopatía inflamatoria idiopática (MII) caracterizada por debilidad muscular subaguda, proximal y simétrica, con evidencia histológica de mionecrosis, escaso infiltrado linfocítico y sin atrofia perifascicular. La detección de autoanticuerpos permite diferenciarla de otras MII y conectivopatías que asocian miositis1, y clasificarla (tabla 1)1,2.
Características de las miopatías inflamatorias idiopáticas
| Dermatomiositis/dermatomiositis juvenil | MNIM | Polimiositis | Miositis de superposición | Miositis de cuerpos de inclusión | |||
|---|---|---|---|---|---|---|---|
| Anti-SRP(22-39%) | Anti-HMGCR(26-50%) | Seronegativa(25-40%) | |||||
| Debilidad muscular | Proximal | Proximal | Proximal | Proximal | Distal (flexores largos de los dedos, extensores de rodilla) | ||
| Manifestaciones extramusculares | Cutáneas (eritema en heliotropo, pápulas de Gottron)Disfagia | Disfagia, afectación pulmonar y cardíaca | Infrecuentes | Frecuentes.Asociación a conectivopatías y malignidad (adultos) | Infrecuentes (salvo disfagia) | Síndrome antisintetasa: enfermedad pulmonar intersticial, manos de mecánico, artritis, síndrome de Raynaud.EsclerodermiaLupus eritematoso sistémico | Infrecuentes (salvo disfagia) |
| Autoanticuerpos | Mi-2, MDA2, TIF-1γ, NXP2, SAE | SRP | HMGCR | Negativos | Inespecíficos | Asociados a síndrome antisintetasa: Jo-1, PL-7, PL-12, HA, EJ, KS, Zo, OJ.Otros: Ku, Ro/SS-S, SS-B, PM/Scl, U-snRNP | c-N1A |
| Histología | Inflamación perimisial, atrofia perifascicular, depósito de HLA-I y complemento en los capilares o sarcolema | Intensa necrosis, depósito de HLA-I y complemento en los capilares o sarcolema | Células T CD8+ endomisiales | Necrosis perifascicular, depósito de HLA-I y II en sarcolema | Células T CD8+ endomisiales, HLA-I, amiloide, vacuolas, tubulofilamentos | ||
Fuente: modificada de Schmidt J.1.
El comienzo en niños es excepcional, especialmente en el subtipo seronegativo, con un único caso reportado3. La ausencia de tratamiento estandarizado en pediatría obliga a extrapolar el abordaje terapéutico del adulto, inicialmente con corticoides e inmunoglobulinas intravenosas (IGIV). Aunque la respuesta suele ser adecuada en niños3, en casos graves es preciso recurrir a otros inmunomoduladores, no existiendo consenso respecto al fármaco de elección.
Presentamos el caso de un niño con MNIM refractaria al tratamiento, que desarrolló afectación cardiaca y posteriormente cutánea compatible con síndrome de superposición (SS) con esclerodermia.
Niño de 10 años con debilidad progresiva, simétrica y no fluctuante de miembros superiores e inferiores e hipotonía axial de 2 meses de evolución. Xerosis e hiperpigmentación en región preesternal, hipogastrio y rodillas e hipopigmentación periocular y perioral (figs. 1a-c). Niega disfagia, fiebre u otros síntomas. En la exploración, debilidad muscular grave, predominantemente axial (escala CMAS 12/52 puntos). Analíticamente: CK 13.798U/l, aldolasa 319U/l, LDH 1.700U/l, GOT/GPT: 509/664U/l, VSG 8mm. Autoanticuerpos, incluyendo anti-SRP y anti-HMGCR, negativos. Afectación miopática predominantemente proximal en EMG. En la RM, extenso edema muscular simétrico de cinturas escapular y pelviana (fig. 1d). Dilatación de 2/3 distales esofágicos, con peristaltismo ineficaz en el tránsito esófago-gástrico-duodenal. El estudio cardiológico (FEVI: 72%) y de función pulmonar resultaron normales. La biopsia muscular evidencia marcada afectación miopática necrosante e intensa sobreexpresión del MCH-I, sin atrofia perifascicular ni depósito de complejo de ataque a la membrana, compatible con MNIM.
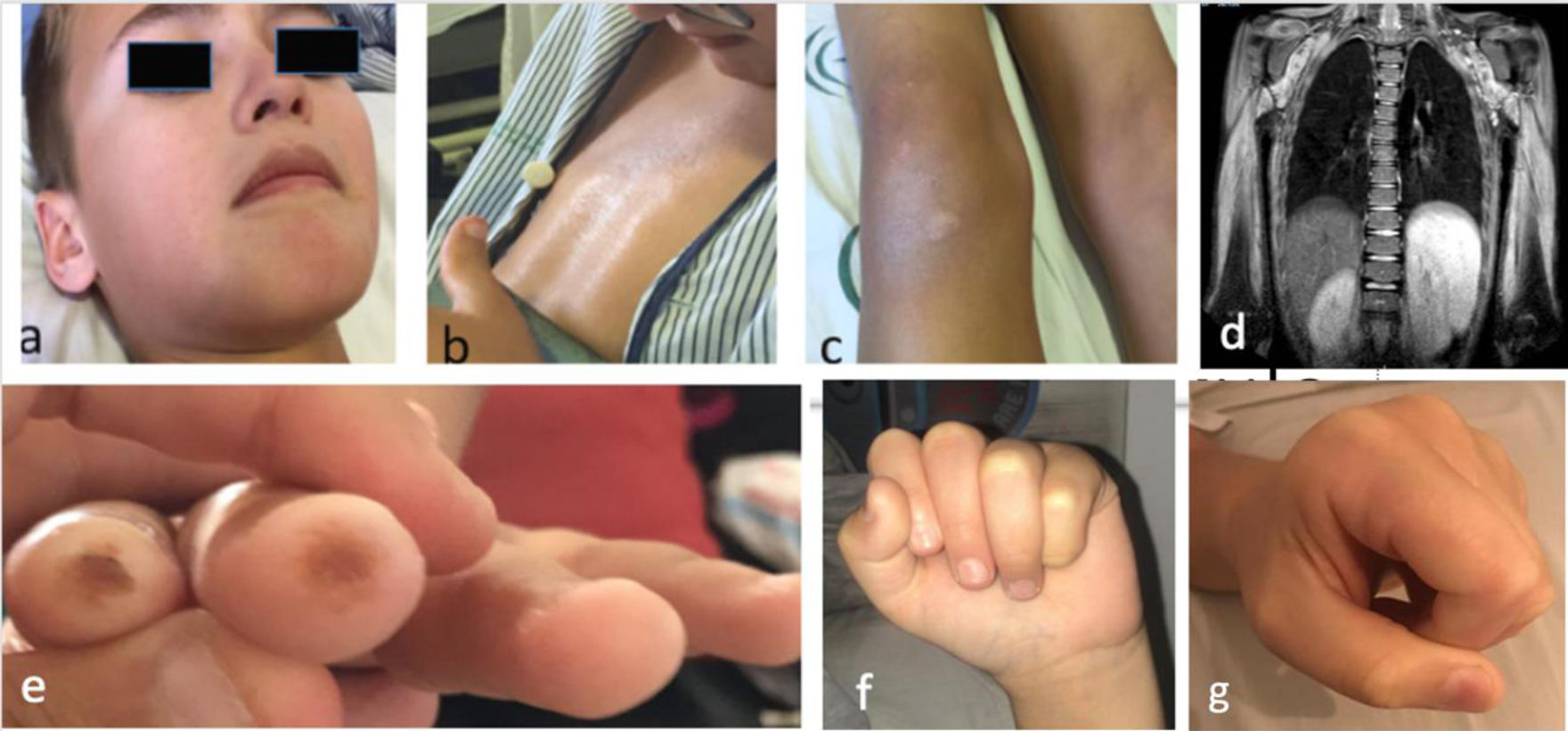
a) Hipopigmentación facial (periocular y perioral); b) Piel de aspecto atrófico, brillante e hiperpigmentada a nivel esternal; c) Hiperpigmentación y lesiones descamativas, de aspecto psoriasiforme, en rodillas; d) Corte coronal de RM (extensa afectación de la musculatura de ambos hombros, tórax y proximal de brazos, bilateral y simétrica, hiperintensa en secuencia STIR indicativo de edema); e) Úlceras digitales; f) Esclerosis cutánea en manos con dificultad para realizar el puño (visión frontal); g) Esclerosis cutánea en manos con dificultad para realizar el puño (visión lateral).
Recibe bolos de metilprednisolona (30mg/kg/día) durante 5 días, IGIV (2g/kg) y metotrexato subcutáneo (15mg/m2/sem). Al alta, leve mejoría clínica con descenso marcado de CK. Tras 48h reingresa por insuficiencia cardiaca con disfunción ventricular (FEVI: 25-30%) y leve derrame pericárdico (1cm), CK 2.500U/l, CK-MB 540ng/ml y troponina 5,94ng/ml. El ECG con ritmo sinusal y elevación de ST y ondas R pequeñas en V4-5, compatibles con daño miocárdico. Ingresa en la UCIP por shock cardiogénico secundario a miopericarditis para soporte inicial con milrinona, cambiando a dopamina por hipotensión, seguido de ciclo de levosimendán. Se optimiza tratamiento de base con bolos de metilprednisolona e inicio de tacrolimus (0,1mg/kg/día), mejorando la función miocárdica hasta normalizarse. El estudio cardiológico ampliado, realizado tras mejoría de la función cardíaca (RM cardíaca y Holter) descarta anomalías estructurales, funcionales, del tejido miocárdico o trastornos del ritmo residuales.
Tras 6 meses de tratamiento médico y rehabilitación, presenta evolución favorable de la debilidad (CMAS 52/52 puntos) y normalización analítica.
Transcurridos 15 meses del inicio, desarrolla úlceras cutáneas en pulpejos de los dedos y endurecimiento cutáneo en manos (figs. 1e-g), realizándose nuevo despistaje de conectivopatía (autoanticuerpos, estudio cardiológico, pulmonar y digestivo) sin alteraciones. Ante la sospecha de SS (esclerodermia y MNIM) se sustituye metotrexato por micofenolato, manteniendo tacrolimus.
Actualmente permanece asintomático, salvo por la esclerosis cutánea que persiste estable.
Consideramos excepcional este caso por la dificultad de afrontar el pronóstico y tratamiento de una entidad tan infrecuente, más aún cuando existen complicaciones con compromiso vital como la afectación cardiaca. Aunque esta se describe en la MNIM, ha sido reportada en formas anti-SRP+, no así en las seronegativas. Por ello, es fundamental evaluar dicha afectación al comienzo y durante el seguimiento, ya que se puede presentar en cualquier momento evolutivo, sin asociar necesariamente debilidad de musculatura periférica4. Además, la aparición de lesiones cutáneas esclerodermiformes obligó a replantear el diagnóstico inicial por el de SS con miopericarditis, dada la mayor frecuencia de afectación miocárdica reportada en estos casos4, aún sin perfil de autoanticuerpos compatible.
La combinación de corticoides, IGIV y metotrexato suele emplearse como tratamiento de primera línea, aunque no existe consenso. Sin embargo, la debilidad en la MNIM suele ser, comparativamente con otras MII, más persistente y resistente al tratamiento, obligando en ocasiones a emplear combinaciones de fármacos5,6, como ocurrió en nuestro paciente. En estos casos tacrolimus, micofenolato, azatioprina, ciclosporina o ciclofosfamida podrían valorarse como tratamientos de segunda línea, reservando rituximab para casos con insuficiente respuesta6. En caso de afectación cardiaca, el soporte con inotrópicos, como levosimendán, permite mantener una aceptable función miocárdica en espera de alcanzar niveles terapéuticos de los inmunomoduladores que conseguirán el control de la enfermedad. Aunque no está establecida la duración del tratamiento, la agresividad del cuadro obliga a plantear la posibilidad de mantenerlo indefinidamente.
Presentaciones previas en congresos: 6.° Foro de la Sociedad Española de Reumatología Pediátrica (SERPE). Murcia, noviembre 2016.
