

La NCG es una entidad heterogénea que se manifiesta en edades precoces de la vida y se caracteriza por un fallo primario en la mielopoyesis con un RAN < 0,5 x 109/l, infecciones graves y riesgo de transformación leucémica.
ObjetivoConocer el curso evolutivo a largo plazo de los pacientes diagnosticados de NCG.
Material y métodosSe analizaron las características clínicas, los métodos diagnósticos, el tratamiento y la evolución de 11 pacientes afectados de NCG.
ResultadosLa mediana de edad al diagnóstico fue de 4 meses (rango: 3 días-12 años). Clínica inicial en todos los casos: infección grave. Mediana de RAN al diagnóstico: 0,2 x 109/l (rango: 0-0,37). El aspirado de médula ósea mostró en todos los casos stop madurativo a nivel de promielocito. El estudio genético mostró en 3 mutaciones, 2 que afectaban al gen ELA2 y una en el gen G6PC3. El G-CSF fue el tratamiento de elección en 9 pacientes. Seis presentaron una buena respuesta a las dosis de entre 5-15 μg/kg/día administrado de 3 a 7días por semana. Tres no respondieron a G-CSF, indicándose un TPH alogénico. En 2 pacientes el TPH fue el tratamiento de primera elección. Mediana de seguimiento de 5 años (rango: 1-10), con una supervivencia del 100% sin ningún caso de transformación leucémica.
ConclusionesA la vista de los datos podemos concluir que el estudio genético de la NCG es útil para establecer una correlación entre genotipo y fenotipo. El tratamiento de elección es la administración G-CSF por vía subcutánea, al que responden las dos terceras partes de los pacientes, indicándose el TPH para los casos de mala respuesta o en aquellos que evolucionan a SMD o leucemia, por lo que el seguimiento de esta entidad es fundamental.
Severe congenital neutropenia (SCN), a heterogeneous condition with onset at early ages, is characterised by primary myelopoiesis failure with an absolute neutrophil count (ANC) < 0.5 x109/L, severe infections and risk of leukaemic transformation.
ObjectiveThe aim of the study was to ascertain the long term outcome of patients with SCN.
Material and methodsThe clinical features, diagnostic methods, treatment and outcome of 11 patients with SCN were analysed.
ResultsThe median age at diagnosis was 4 months (range: 3 days-12 years). The primary clinical manifestation was severe infection. Median ANC at diagnosis: 0.2 x 109/L (range: 0-0.37). Bone marrow aspirate showed maturation arrest at promyelocyte stage in all cases. Genetic studies revealed 3 mutations, two in ELA-2 gene and 1 in G6PC3 gene, showing a correlation between genotype and phenotype. Granulocyte Colony Stimulating Factor (G-CSF) was the first-line treatment in 9 patients; six of whom showed a good response at doses between 5 and 15 μg/kg/day for 3-7 days/week. The remaining 3 patients failed to respond to G-CSF and allogeneic stem cell transplantation (SCT) was indicated. Furthermore, SCT was the treatment of choice in two cases. Median follow-up of the cohort was 5 years (range: 1-10 years) with 100% survival and no cases of leukaemic transformation.
ConclusionsWe conclude that genetic study is useful for establishing a correlation between genotype and phenotype. The treatment of choice for SCN is G-CSF to which 2/3 of patients should respond; while SCT is reserved for cases of poor response or those evolving to myelodysplastic syndrome (MDS) or leukaemia; thus close follow-up of this condition is essential.
La neutropenia congénita grave (NCG) es una entidad heterogénea que engloba una serie de síndromes cuya característica común es un recuento absoluto de neutrófilos (RAN) inferior a 0,5 x 109/l debido a un fallo primario en la mielopoyesis. Se manifiesta en forma de infecciones bacterianas desde edades muy tempranas1-3. A pesar de que los mecanismos fisiopatológicos no han sido aclarados todavía, se sabe que la alteración cuantitativa de los neutrófilos se fundamenta en una alteración cualitativa que tiene como resultado un acortamiento de su vida media3.
Clínicamente se manifiesta en forma de infecciones desde los primeros meses de vida, requiriendo en la mayoría de los casos ingreso hospitalario para la administración de antibióticos por vía intravenosa.
El tratamiento de primera elección es la administración de factor estimulador de colonias de granulocitos (G-CSF), reservándose el trasplante de progenitores hematopoyéticos (TPH) para los casos de mala respuesta o de transformación leucémica2.
Pacientes y métodosSe analizaron las características clínicas, los métodos diagnósticos, la evolución y el tratamiento de los pacientes diagnosticados de NCG en nuestro centro en los últimos 28 años. Se incluyó en el diagnóstico de NCG a aquellos pacientes con un RAN inferior a 0,5 x 109/l de origen central y primario.
A todos los pacientes se les realizó inicialmente un hemograma y un aspirado de médula ósea, con estudio morfológico y citogenético. En 10 pacientes se realizó un estudio genético para los genes ELA2 y HAX1; en los varones se estudió el gen WASP y en los que presentaban un síndrome polimalformativo característico, el gen G6PC3. El estudio de alteraciones genéticas en el dominio externo del receptor de G-CSF se realizó en los sujetos en que no se obtuvo respuesta al tratamiento con G-CSF.
Se realizó un registro de los eventos infecciosos que requirieron ingreso hospitalario y administración de antibióticos por vía intravenosa.
El seguimiento clínico y hematológico se realizó de forma periódica y tan frecuentemente como fue necesario. El estudio morfológico y citogenético de la médula ósea se realizó anualmente de forma rutinaria. La valoración de la densidad ósea se determinó anualmente mediante radiología simple y densitometría.
ResultadosOnce pacientes, 7 niños y 4 niñas, fueron diagnosticados de NCG. En ningún caso constaban antecedentes familiares de NCG, muertes precoces o síndromes polimalformativos.
La mayoría de los pacientes se diagnosticaron dentro del primer año de vida (72%), con una edad mediana al diagnóstico de 4 meses (rango: 3 días-12 años). En todos los casos la manifestación inicial fue una infección bacteriana que requirió administración de antibiótico de amplio espectro por vía intravenosa. En 6 casos se presentó en forma de infecciones cutáneas en los primeros meses de vida (4 abscesos y 2 ectimas gangrenosos), 3 pacientes presentaron septicemia y 2 neumonía.
En 4 pacientes la neutropenia se asoció a otras anomalías constitucionales: retraso mental no filiado (2 casos), persistencia de ductus arterioso (1 caso) y síndrome polimalformativo (1 caso). Este último era un paciente de 11 años, de raza árabe, con antecedentes de infecciones de repetición desde la primera infancia, que presentaba neutropenia grave asociada a cardiopatía, criptorquidia, bronquiectasias y alteración de la vascularización periférica, en el que pudo demostrarse una mutación en el gen G6PC3.
En todos los casos la cifra de leucocitos totales al diagnóstico ajustada por edad y sexo fue normal. La mediana de RAN fue de 0,2 x 109/l (rango: 0-0,37). En 8 pacientes se acompañó de monocitosis y en 6 de eosinofilia. La serie roja y la serie plaquetar fueron normales en todos los casos, excepto en el caso de la mutación en el gen G6PC3, en el que se detectó una trombocitopenia leve/moderada.
El estudio de médula ósea mostró detención de la maduración de la serie blanca a nivel de promielocito/mielocito. En un caso se detectó monosomía del cromosoma 7.
Se detectaron 2 casos con una mutación en el gen ELA2, uno de ellos con neutropenia cíclica, y en el paciente que asociaba un síndrome polimalformativo se halló una mutación en el gen G6PC3.
El primer paciente de nuestra serie fue diagnosticado en la era pre-GCSF. Inicialmente, se administró GM-CSF, sin obtenerse respuesta, por lo que se indicó un TPH de médula ósea de hermano HLA con muy buena evolución posterior.
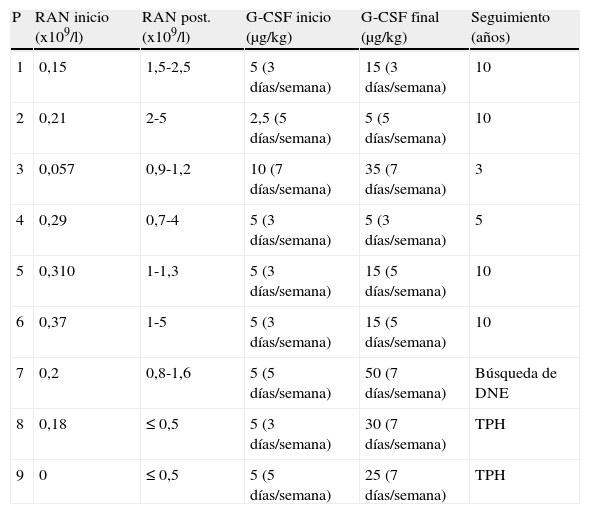
En 9 pacientes se indicó administración por vía subcutánea de G-CSF, con una dosis inicial de 2,5-5 μg/kg/día (3-7 días/semana) y un ajuste de dosis progresivo hasta lograr un buen control clínico y analítico. Seis de estos pacientes presentaron una buena respuesta con una dosis media de 5-15 μg/kg /día (3-7 días/semana), con mejoría clínica significativa y RAN entre 0,7-1,2 x 109/l. En 3 pacientes el recuento de neutrófilos no superó los 0,5 x 109/l. Se estudió la posibilidad de que la falta de respuesta se asociara a alteraciones genéticas en el receptor de G-CSF, sin encontrarse en ningún caso. En 2 de ellos se realizó un TPH de sangre de cordón umbilical (SCU) de donante no emparentado (DNE) con buena respuesta. El tercer paciente que no respondió a G-CSF se encuentra en búsqueda de donante no emparentado HLA compatible. Las características del tratamiento que recibió cada paciente se muestran en la tabla 1.
Recuento de neutrófilos y tratamiento con G-CSF
| P | RAN inicio (x109/l) | RAN post. (x109/l) | G-CSF inicio (μg/kg) | G-CSF final (μg/kg) | Seguimiento (años) |
| 1 | 0,15 | 1,5-2,5 | 5 (3 días/semana) | 15 (3 días/semana) | 10 |
| 2 | 0,21 | 2-5 | 2,5 (5 días/semana) | 5 (5 días/semana) | 10 |
| 3 | 0,057 | 0,9-1,2 | 10 (7 días/semana) | 35 (7 días/semana) | 3 |
| 4 | 0,29 | 0,7-4 | 5 (3 días/semana) | 5 (3 días/semana) | 5 |
| 5 | 0,310 | 1-1,3 | 5 (3 días/semana) | 15 (5 días/semana) | 10 |
| 6 | 0,37 | 1-5 | 5 (3 días/semana) | 15 (5 días/semana) | 10 |
| 7 | 0,2 | 0,8-1,6 | 5 (5 días/semana) | 50 (7 días/semana) | Búsqueda de DNE |
| 8 | 0,18 | ≤ 0,5 | 5 (3 días/semana) | 30 (7 días/semana) | TPH |
| 9 | 0 | ≤ 0,5 | 5 (5 días/semana) | 25 (7 días/semana) | TPH |
DNE: donante no emparentado; G-CSF final: dosis final de factor estimulador de colonias de granulocitos; G-CSF inicio: dosis inicial de factor estimulador de colonias de granulocitos; RAN: recuento absoluto de neutrófilos; RAN post.: recuento absoluto de neutrófilos posterior al tratamiento; TPH: trasplante de progenitores hematopoyéticos.
En el paciente que presentó monosomía del cromosoma 7 se indicó un TPH alogénico como terapia de primera línea. Se realizó un trasplante de SCU de DNE, con recuperación de la neutropenia.Se analizaron un total de 66 eventos infecciosos, que requirieron ingreso y tratamiento antibiótico por vía intravenoso. En un 37% se trataba de infecciones cutáneas y de partes blandas, y en un 30% neumonías, destacando 6 casos de neumonía necrotizante de evolución tórpida. La media de ingreso hospitalario fue de 10 días. En todos los casos se administró antibioterapia de amplio espectro de forma inicial, ajustada tras los resultados microbiológicos. En 31 casos se pudo identificar el germen responsable, destacando Pseudomonas aureginosa (6 casos), Escherichia coli (4 casos) y Salmonella typhimorium (2 casos). Once casos precisaron cura o drenaje quirúrgico y 2 pacientes requirieron ingreso en la UCI-P. La supervivencia fue del 100%. La tasa de ingreso por infección fue de entre 3-4 por paciente/año hasta lograr un tratamiento efectivo. En los pacientes que presentaron una buena respuesta a G-CSF, la tasa disminuyó a 0,075 ingresos por infección/paciente/año. La figura 1 muestra los eventos infecciosos registrados.

Con una mediana de seguimiento de 5 años, en ningún caso se detectó transformación a síndrome mielodisplásico ni leucemia.
En 2 pacientes, tras 6 y 8 años, respectivamente, de tratamiento diario con GCSF, se detectó osteoporosis por lo que precisó tratamiento con calcio y bifosfonatos.
DiscusiónLa NCG es una entidad poco frecuente y que representa una gran variedad de alteraciones genéticas, manifestaciones clínicas y respuesta al tratamiento. Aparece en edades precoces de la vida y debe tenerse en cuenta ante el diagnóstico de infecciones de repetición3-5.
Fue descrita inicialmente por Kostmann (1956) como una enfermedad de herencia autosómica recesiva; posteriormente, se atribuyó este síndrome a mutaciones en el gen HAX11. El avance en el conocimiento de esta entidad ha permitido describir formas de herencia autosómica dominante y otras formas de herencia autosómica recesiva6-8. Los estudios de ADN realizados describen mutaciones del gen de la elastasa 2 (ELA 2) como la causa más frecuente de NCG, con un patrón de herencia autosómica dominante1. En las formas recesivas se han identificado además de mutaciones en el gen HAX1, otras en los genes p14, G6PC3, TAZ y GASP. Sin embargo, en aproximadamente un 25% de los casos no se puede identificar ninguna alteración genética, según los datos reportados por el Registro Internacional de NCG (SCNIR)4. En nuestra serie se realizó el estudio genético a 10 pacientes, encontrándose únicamente 3 mutaciones: dos en el gen ELA2 y 1 en G6PC3.
La presentación clínica inicial son las infecciones de repetición, sobre todo aquellas que aparecen durante los primeros meses de vida en forma de fiebre sin foco, infecciones cutáneas como ectimas o abscesos e infecciones respiratorias1. El diagnóstico inicial en nuestra serie de casos se corresponde con estos hallazgos clínicos descritos en la literatura. En ocasiones puede acompañarse de otras manifestaciones constitucionales, como alteraciones del sistema nervioso central, malformaciones orgánicas, gingivitis y caída dentaria precoz. Muchas de estas manifestaciones se corresponden con una anomalía genética específica responsable del síndrome3. Mutaciones en el gen HAX1 asocian con frecuencia neutropenia, retraso mental y convulsiones6, mientras que otras mutaciones en el gen G6PC3 asocian neutropenia un síndrome polimalformativo con cardiopatía, criptorquidia, alteraciones cutáneas y, de forma típica, vascularización periférica marcada9,10, como era el caso del paciente descrito en nuestra serie.
El diagnóstico de neutropenia de origen central se establece mediante el estudio de médula ósea, que muestra un stop madurativo a nivel de promielocio/mielocito. Sin embargo, la existencia en algunos casos de una correlación entre genotipo y fenotipo sugiere la realización de un estudio genético dirigido de NCG para tipificación de la misma3,8.
El G-CSF subcutáneo es el tratamiento de elección, consiguiendo en la mayoría de los casos una buena respuesta clínica con una disminución significativa de los procesos infecciosos y un RAN ≥ 0,5 x 109/l2,11. En nuestra serie, 9 de los 11 pacientes estudiados recibieron G-CSF como primer tratamiento, 6 de los cuales presentaron buena respuesta, disminuyendo de forma muy significativa el número de ingresos hospitalarios por infección. Entre un 10 y un 40% de los pacientes, según las series consultadas4, no responden a G-CSF. En una minoría de pacientes la resistencia a G-CSF puede deberse a una mutación en el dominio externo del receptor de G-CSF8. Tres de nuestros pacientes no respondieron a la administración de G-CSF, por lo que tuvo que indicarse la realización de un TPH. En ninguno de ellos se detectó la mutación del receptor de G-CSF.
Entre los efectos secundarios de este tratamiento destaca la aparición de osteopenia/osteoporosis que suelen aparecer a medio/largo plazo4,11, por lo que es importante monitorizarlos y establecer un diagnóstico y un tratamiento tempranos. En nuestra serie, durante el seguimiento, se realizó una densitometría anual para descartar dichas alteraciones. Hasta el momento actual, 2 pacientes presentan alteraciones óseas, que aparecieron a los 6 y 8 años, respectivamente, del inicio de tratamiento con G-CSF.
La NCG es una entidad con riesgo de evolucionar hacia una leucemia aguda, siendo más frecuente la leucemia mieloide aguda (LAM)12,13. La incidencia acumulada de transformación leucémica, según los datos reportados por el SCNIR, es del 21% a los 10 años de tratamiento continuado con G-CSF4,14,15. En ocasiones, el estudio citogenético muestra alteraciones cromosómicas, en especial la monosomía del cromosoma 7, que puede preceder al diagnóstico de LAM. En nuestra serie, en 1 paciente se detectó una monosomía del cromosoma 7, motivo por el cual se realizó un trasplante de progenitores hematopoyéticos como tratamiento de primera elección.
Estudios recientes han descrito otro tipo de mutación del receptor de G-CSF (CSF3R), distinta de la que afecta al dominio externo, asociada a la resistencia al tratamiento con G-CSF, que afecta al dominio intracitoplasmático1,8,15,16. Dicha mutación aparece en los pacientes seguidos a largo plazo y, aunque el mecanismo fisiopatológico es controvertido, se asocia a un mayor riesgo de desarrollo de síndrome mielodisplásico o LAM8,17,18. Si el tratamiento continuado con G-CSF puede contribuir a la malignización o es simplemente la evolución natural de la enfermedad no está suficientemente aclarado.
En la NCG el riesgo de evolucionar hacia un proceso hematológico maligno hace necesario el seguimiento a largo plazo, con la realización periódica de estudios morfológicos, citogenéticos y moleculares en la médula ósea que permitan una detección precoz de alteraciones citogenéticas con riesgo de malignización, síndrome mielodisplásico o transformación leucémica. De acuerdo con la literatura y con nuestra experiencia, estos estudios deberían realizarse de forma anual, siempre que exista una estabilidad clínica y analítica, debiéndose adelantar ante manifestaciones clínicas o cambios en el hemograma que alerten de una posible transformación maligna.
En los pacientes que no responden a G-CSF y en aquellos que presenten una alteración preleucémica o una entidad maligna establecida está indicada la realización de un TPH alogénico3,12,19. En nuestra serie de casos se indicó el TPH a 5 pacientes: en 3 pacientes por no responder al tratamiento con G-CSF; en 1 caso por la aparición de monosomía en el cromosoma 7 y en otro por diagnosticarse antes del uso del G-CSF como tratamiento en la NCG. Todos ellos presentaron una buena evolución tras el TPH.
ConclusionesLa NCG es una entidad heterogénea en cuanto a la etiología, la fisiopatología, la clínica y la respuesta al tratamiento. Debe sospecharse ante los cuadros de infecciones bacterianas graves que acontecen en edades precoces. El diagnóstico se establece tras el estudio de médula ósea que muestra stop madurativo a nivel de promielocito/mielocito. Puede acompañarse de otras anomalías constitucionales; el estudio genético en ocasiones pone en relación el genotipo con el fenotipo. El tratamiento de primera elección es el G-CSF, al que responden los dos tercios de los pacientes, logrando una disminución significativa del número de infecciones graves. El trasplante alogénico de progenitores se reserva para los pacientes que no responden a G-CSF o bien para aquellos casos en los que aparezca transformación leucémica o signos de evolución hacia la malignidad, como síndrome mielodisplásico y algunas alteraciones citogenéticas. El riesgo de presentar infecciones de repetición potencialmente graves, transformación hacia una entidad maligna y osteoporosis con el tratamiento con G-CSF hacen necesario el seguimiento a medio y largo plazo de estos pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
