



La enfermedad de Hirschsprung está causada por un defecto de la migración celular desde la cresta neural hasta el tracto gastrointestinal, resultando en la ausencia de neuronas en el plexo mientérico. Mutaciones en varios genes han sido asociadas a la enfermedad de Hirschsprung, la mayoría afectando a la vía del protooncogén RET. El objetivo de este estudio es la descripción de mutaciones tanto descritas como nuevas asociadas a la enfermedad de Hirschsprung, así como sus implicaciones pronósticas.
Material y métodosAnálisis retrospectivo de pacientes con enfermedad de Hirschsprung y resultados genéticos positivos desde 1970 hasta 2013.
ResultadosEn la serie global, 21 pacientes tenían resultados genéticos positivos, 17 de ellos afectando la vía del protooncogén RET. Dos de las mutaciones son nuevas y no han sido previamente descritas en la literatura médica.
ConclusionesEl protooncogén RET es el principal gen asociado a la enfermedad de Hirschsprung. Todavía hay múltiples mutaciones desconocidas relacionadas con la patogenia de la enfermedad. El estudio genético del gen RET debe formar parte del estudio diagnóstico de todos los pacientes con enfermedad de Hirschsprung, así como de sus familiares de primer grado en caso de que las mutaciones estén asociadas a los síndromes MEN2A y MEN2B.
Hirschsprung disease is caused by an impairment in cell migration from the neural crest to the gastrointestinal tract, resulting in an absence of neurons in the myenteric plexus. Many mutations in several genes have been associated to Hirschsprung disease; most of them affecting the RET proto-oncogen pathway. The purpose of this study is the description of novel and known mutations in genes associated to Hirschsprung disease and their prognostic implications.
Material and methodsRetrospective analysis of patients with Hirschsprung disease and positive genetic studies evaluated from 1970 to 2013.
ResultsWe found 21 positive genetic studies in the global series, 17 of them involving the RET proto-oncogene. Two of the mutations are novel and they have not been reported in the medical literature.
ConclusionsThe RET protooncogene is the main gene associated with Hirschsprung disease. There are still multiple unknown mutations related to the pathogenesis of the disease. The study of this gene must be part of the work-up of all patients with Hirschsprung disease, as well as their first degree relatives if the mutation is associated with MEN2A and MEN2B syndromes.
La enfermedad de Hirschsprung (EH) está causada por un defecto de la migración celular desde la cresta neural hasta el tracto gastrointestinal, resultando en la ausencia de neuronas en el plexo mientérico1,2. La incidencia global de la EH es de aproximadamente un caso por 5.000 recién nacidos3, y es mayor en la población asiática, seguida de la afroamericana y la europea. En aproximadamente el 80% de los pacientes la enfermedad se manifiesta en la forma de segmento corto (o rectosigmoidea). En el 15% de los afectados, el aganglionismo se extiende proximalmente, resultando en la forma de segmento largo. En el 5% restante el defecto se extiende a la totalidad del colon4. Se dan, asimismo, casos poco frecuentes en los que la afectación alcanza el intestino delgado. El cuadro normalmente comienza con dismotilidad intestinal, manifestándose con obstrucción intestinal, distensión, estreñimiento o ausencia de eliminación de meconio. El diagnóstico se confirma mediante estudio histológico y se basa en la presencia o ausencia de células ganglionares en el plexo mientérico. El tratamiento es quirúrgico y consiste en la resección del segmento afectado, seguido de descenso endorrectal del colon gangliónico y anastomosis terminoterminal5,6.
Se han descrito varias mutaciones genéticas asociadas a la EH7. La identificación de causas genéticas es más frecuente en casos de enfermedad de segmento largo en comparación con los de segmento corto8,9. En múltiples familias con EH se han identificado mutaciones en el gen del receptor transmembrana de la tirosina-cinasa (RET), una región que actualmente se conoce como un locus de susceptibilidad para la EH1 (MIM 142623). Se han identificado otros loci asociados a la EH aislada. Por ejemplo, la EH2 (MIM 600155) se asocia a mutaciones en el gen EDNRB, localizado en el cromosoma 13q22; la EH3 (MIM 613711), a mutaciones en el gen GDNF, localizado en el cromosoma 5p13; la EH4 (MIM 613712), a mutaciones en el gen EDN3, en el 20q13; la EH5 (MIM 600156) se asocia a mutaciones localizadas en el 9q31; la EH6 (MIM 606874), a mutaciones en el 3p21; la EH7 (MIM 606875), a mutaciones en el 19q12; la EH8 (MIM 608462), a mutaciones en el 16q23, y la EH9 (MIM 611644), a mutaciones en el 4q31-q32. Por otro lado, hay pacientes en los que la EH se manifiesta en el contexto de un síndrome10, como por ejemplo el síndrome de Bardet-Biedl, el síndrome de Down11, el síndrome de Fryns, el síndrome de Goldberg-Shprintzen, el síndrome de hipoventilación central congénita12, la displasia intestinal neuronal tipo B, el síndrome de Mowat-Wilson13, el síndrome MEN2A, el síndrome MEN2B14, la neurofibromatosis tipo 1, el síndrome de Pitt-Hopkins, el síndrome de Smith-Lemli-Opitz o el síndrome de Waardenburg tipo 4. Por añadidura, en aproximadamente el 18% de los pacientes, la EH se asocia a malformaciones congénitas15, en su mayoría cardiopatías (tales como comunicación interauricular, comunicación interventricular, ductus arterioso, tetralogía de Fallot), malformaciones gastrointestinales16 (malrotación intestinal, ano imperforado, divertículo de Meckel, fístula anorrectal), malformaciones genitourinarias17 (criptorquidia, hernia inguinal, hipospadias, malformación renal, fístula uretral) y malformaciones del sistema nervioso central (discapacidad intelectual, malformación de Dandy-Walker, microcefalia).
El estudio tenía por objetivo describir las mutaciones encontradas en pacientes con EH seguidos en un hospital terciario objeto de estudio genético completo, establecer si habían sido descritas previamente y considerar la relevancia clínica y las implicaciones pronósticas del estudio genético en el manejo de la EH18.
MétodosAnálisis retrospectivo de las historias clínicas de pacientes con EH diagnosticados entre 1970 y 2013. Se incluyó a pacientes con confirmación histológica de la enfermedad, es decir, en los que se evidenció la ausencia de células ganglionares en el plexo mientérico en la biopsia rectal. La hiperplasia de fibras nerviosas colinérgicas, el incremento en la actividad de la acetilcolinesterasa en la muscularis mucosae o la inmunorreactividad reducida a la calretinina en la lámina propia se consideraron hallazgos histológicos que apoyaban el diagnóstico pero no eran confirmatorios.
El estudio se realizó en conformidad con los principios éticos de la Declaración de Helsinki (revisión de Tokio, octubre 2004) y con las regulaciones concernientes a la confidencialidad de los pacientes. Se obtuvo la aprobación del Comité Ético del hospital antes de comenzar el estudio, así como el consentimiento informado de los padres de los 3 pacientes incluidos en el análisis.
El estudio se centró en aquellos pacientes cuyas historias clínicas contenían resultados genéticos positivos registrados en la historia clínica. Se recogieron datos clínicos y epidemiológicos y se compararon con los resultados del estudio genético, con énfasis en las mutaciones nuevas.
Se compararon las mutaciones detectadas en los pacientes con las descritas en la literatura médica y en las siguientes bases de datos de genética: Domain Mapping of Disease Mutations (DMDM), BioMuta 2.0, UniProt, COSMIC, Clinical Variants (ClinVar), Leiden Open source Variation Database (LOVD) y Exome Aggregation Consortium (EXAC)
Los métodos empleados en el estudio genético variaron durante el período de estudio. De 1970 a 2007, la única prueba realizada fue el cariotipo. Entre 2007 y 2011, se introdujeron el array de hibridación genómica comparada y el análisis del gen RET. A partir de 2011 se han analizado otros genes (en caso de sospecha), directamente mediante la secuenciación de Sanger o mediante paneles para la detección de enfermedades mendelianas (secuenciación del genoma clínico).
Se realizaron análisis genéticos de ADN en muestras de linfocitos de sangre periférica mediante la amplificación por reacción en cadena de la polimerasa en cadena y posterior secuenciación de las regiones codificantes y las uniones intrón-exón del gen RET. El análisis de las secuencias codificantes y las uniones intrón-exón del gen RET (NM_020975.4) se llevó a cabo mediante secuenciación directa. Se diseñaron cebadores con ayuda de las aplicaciones de software Primer 3 v0.4.0 (http://bioinfo.ut.ee/primer3-0.4.0/) y SNPCheck V3 (https://secure.ngrl.org.uk/SNPCheck/snpcheck.htm). Los productos de la reacción en cadena de la polimerasa se secuenciaron con el BigDye Terminator Cycle Sequencing Kit (Applied Biosystems; Foster City, California, EE. UU.) en un secuenciador ABI3730XL (Applied Biosystems; Foster City, California, EE. UU.). La conservación, la predicción de patogenicidad in silico y el análisis de frecuencia en población de control (Exome Aggregation Consortium) de las variantes de RET identificadas se realizó con el software Alamut V2.6-1 (Interactive Biosoftware; Ruan, Francia) y MutPred (http://mutpred.mutdb.org/). También consultamos la base de datos de mutaciones en el gen RET19 para comprobar si alguna de las variantes detectadas había sido descrita previamente, lo que apoyaría su papel en la patogenia de la EH.
El estudio de reordenamientos genéticos se realizó mediante cariotipo y/o array de hibridación genómica comparada. El gen paired-like homeobox 2b (PHOX2B) se analizó mediante secuenciación de Sanger o mediante secuenciación del exoma clínico, siguiendo las recomendaciones y guías del American College of Medical Genetics and Genomics20.
ResultadosEn el período de estudio (1970-2013), hubo sospecha clínica de EH en 193 pacientes. Se obtuvo confirmación histológica del diagnóstico de EH en 160 pacientes. La ratio varón:mujer fue de 113:47. La edad al diagnóstico osciló entre los 0 meses y los 7 años.
No encontramos registro de estudios genéticos en la mayoría de los pacientes, probablemente porque no se incluyeron en el protocolo rutinario de diagnóstico de la EH hasta las últimas décadas. El primer registro de estudio genético en un paciente correspondió al año 1989. Se identificó a 25 pacientes con estudio genético, detectándose anomalías en 21.
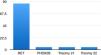
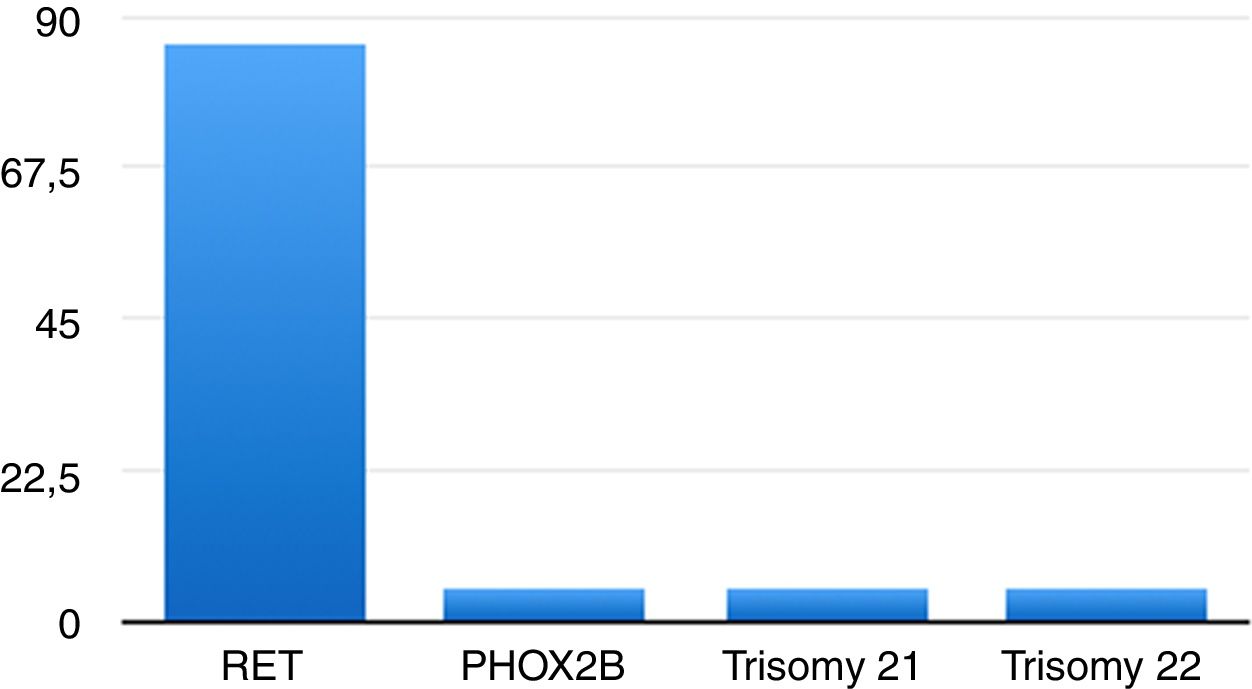
En 17 casos (80,95%) las variantes correspondieron a mutaciones en el protooncogén RET, una de ellas benigna (2608-24 G>A; 4,76%)21, y en un paciente se detectó una mutación en el gen PHOX2B (4,76%)22,23. Dos pacientes tenían trastornos genéticos: uno, trisomía 21 (4,76%), y el otro, trisomía 22 (4,76%) (fig. 1).
De los 17 pacientes con mutaciones en el protooncogén RET, 13 tenían mutaciones descritas previamente en la literatura médica. Dos pacientes tenían una combinación de 2 mutaciones (c.1083C>T;p.R360W y c.215T>C;p.L72P). Por lo tanto, se detectaron un total de 11 mutaciones distintas en nuestros pacientes (tabla 1).
Resultados del estudio genético del gen RET
| Secuencia de la mutación | Descripción |
|---|---|
| c.988C>T;p.R330N | Asociación con EH descrita en la literatura |
| c.107A>G;p.Y36C | Asociación con EH descrita en la literatura |
| c.1083C>T;p.R360W | Asociación con EH descrita en la literatura |
| c.428 C>G;pA143G | Asociación con EH descrita en la literatura |
| 987dupT;pF329FfsX24 | Asociación con EH descrita en la literatura |
| c.1252C>T;p.R418X | Asociación con EH descrita en la literatura |
| c.1859G>A;p.C620Y | Asociación con EH descrita en la literatura |
| G>A;p.D300N | Asociación con EH descrita en la literatura |
| c.1831T>G;p.C611G | Asociación con EH descrita en la literatura |
| c.215T>C;p.L72P | No descrita previamente en la literatura |
| c.367C>T; p.L123F | No descrita previamente en la literatura |
Nueve de las 11 mutaciones detectadas (81,82%) estaban registradas en bases de datos como mutaciones patogénicas o que incrementan el riesgo de desarrollar la enfermedad.
Dos de las 11 mutaciones (18,18%) (c.367 C>T;p.L123F y c.215T>C;p.L72P) no habían sido descritas previamente en la literatura o registradas en bases de datos genéticas.
Hallazgos clínicos en pacientes con mutaciones nuevas y en el paciente con la mutación en el gen PHOX2BPaciente 1 (c.367C>T;p.L123F)Niño nacido pretérmino fruto de embarazo gemelar dicoriónico y biamniótico. Además de los síntomas digestivos, el paciente tenía múltiples comunicaciones interauriculares de pequeño tamaño, que se cerraron espontáneamente antes de alcanzar los 4 años de edad. No había otros antecedentes de interés.
A las 24h de nacer, el paciente había desarrollado distensión abdominal, vómitos y estreñimiento. A los 5 días, recibió diagnóstico de perforación cecal, que se trató con una hemicolectomía derecha con ileostomía distal seguida de reconstrucción con anastomosis ileocólica a los 7 meses de edad. Tras la cirugía, el paciente sufrió varios episodios de obstrucción intestinal, manejados médicamente en un principio hasta que se realizó biopsia para evaluar la obstrucción recurrente. Los hallazgos de la biopsia fueron compatibles con EH en las muestras de recto, colon sigmoide, colon descendente y ángulo esplénico (ausencia de neuronas en el plexo mientérico, aunque sin hiperplasia de las neuronas colinérgicas), confirmándose el diagnóstico de EH de segmento largo. Con base en estos hallazgos, se practicó una colostomía. El estudio genético reveló una mutación en el gen RET (c.367C>T;p.L123F). En consecuencia, se extendió el estudio a los familiares de primer grado. Se detectó la misma mutación en el padre y el abuelo del paciente, ambos asintomáticos.
Un año después, el paciente presentó estreñimiento en la colostomía. Se recogieron biopsias adicionales confirmándose la ausencia de neuronas y se practicó una nueva colostomía 14 cm proximal a la anterior, conectando con una porción sana del colon. A los 3 años de edad, el paciente fue referido a nuestro centro y fue sometido a cirugía electiva mediante el procedimiento de Swenson, realizándose una ileostomía protectora que se cerró 6 meses después. Durante el seguimiento ambulatorio se detectó estenosis de la anastomosis leve, confirmada mediante colonoscopia, tratada con éxito mediante dilataciones.
A pesar de las precauciones mencionadas, el paciente sufrió un episodio grave de enterocolitis a los 4 años de edad. El manejo fue conservador, y el paciente se fue de alta con estimulación rectal e irrigación diarias.
Actualmente el paciente tiene 7 años, va al colegio y presenta el nivel de desarrollo propio de su edad. Su desarrollo físico es adecuado. Ha adquirido control de esfínteres y defeca a diario, aunque aún se somete a un enema cada semana para prevenir posibles episodios de enterocolitis.
Paciente 2 (c.215T>C;p.L72)Niña nacida a término (37 semanas) con peso adecuado para la edad gestacional (2.950g) en parto sin complicaciones. Aparte de la EH, tenía antecedentes de foramen oval permeable. A las 48h de vida la paciente presentó signos y síntomas de obstrucción intestinal (distensión abdominal, vómitos biliosos y retraso en la evacuación del meconio). La obstrucción se resolvió con la evacuación del meconio 36h después. En los meses siguientes, la paciente presentó falta de apetito y malnutrición, a pesar de eliminar heces normales. A los 5 meses de edad presentó fiebre, distensión abdominal y rechazo del alimento. La radiografía abdominal reveló neumoperitoneo y dilatación de las asas intestinales. La paciente requirió intervención quirúrgica, practicándose una ileostomía. Durante el procedimiento se detectó y cerró una perforación cecal. También se obtuvieron muestras para biopsia del recto, el colon sigmoide, descendente, transverso y ascendente, ciego e íleon, encontrándose características compatibles con la EH en todas, lo que confirmó enfermedad de segmento largo. También se obtuvieron muestras del resto del intestino delgado, cuya histología resultó ser normal. El estudio genético detectó una mutación en el gen RET (c.215T>C;p.L72).
A los 22 meses de edad, la paciente fue sometida a cirugía programada de descenso abdominoperineal con técnica de Swenson, practicándose una nueva ileostomía que fue cerrada 5 meses después. En los meses que siguieron a la operación la paciente tuvo episodios ocasionales de vómitos y una ingesta insuficiente debida a intolerancia oral y una ganancia de peso inadecuada, y requirió nutrición enteral continua nocturna en los meses que siguieron. En la evaluación más reciente, la paciente tenía 14 años y presentaba un nivel de desarrollo adecuado para su edad. Debido a su dificultad para tolerar la nutrición oral, continúa bajo seguimiento por el equipo de rehabilitación nutricional.
La hermana menor de esta paciente tenía la misma mutación en el gen RET, heredada de la madre. El estudio genético en el padre no detectó anomalías. Nació a término y evacuó el meconio a las 8h de vida, y no tenía antecedentes de interés. Presentó a las 28h de vida vómitos biliosos y rechazo del alimento. Fue sometida a una laparotomía exploradora, en la que se obtuvieron muestras de biopsia, diagnosticándose EH de segmento largo, en este caso limitada al colon descendente. Esta paciente fue tratada con cirugía electiva, con la misma técnica de descenso abdominoperineal, a los 15 meses. La ileostomía se cerró 4 meses después. Tras la cirugía, la paciente desarrolló una eventración supraumbilical en la línea media por encima de la cicatriz de la laparotomía, que no causó complicaciones y se corrigió mediante cirugía a los 3 años de edad.
Tras la cirugía, el problema principal en esta paciente ha sido la baja ganancia de peso, como ocurrió con su hermana. También ha tenido 2 episodios de enterocolitis, presentando fiebre, vómitos, dolor abdominal y estreñimiento. Ambos episodios se manejaron con tratamiento conservador.
Actualmente, la niña tiene 8 años, presenta un control adecuado de las deposiciones y continúa bajo seguimiento ambulatorio.
Paciente con mutación en el gen PHOX2B (c.367C>T;p.L123F)Niño nacido a término con un peso adecuado para la edad gestacional, fruto de un embarazo sin complicaciones. Aparte de EH, el paciente tenía antecedentes relevantes de nefrolitiasis en el lado derecho, con hidronefrosis secundaria, síndrome de hipoventilación central congénita (hipoventilación nocturna) y alergia a la proteína de la leche de vaca.
A los 12 días de vida el paciente presentó distensión abdominal, náuseas y vómitos. Se realizó diagnóstico de obstrucción intestinal completa, requiriendo laparotomía urgente con realización de colostomía a nivel del ángulo hepático. No hubo complicaciones en el postoperatorio inmediato y el paciente fue dado de alta con la colostomía. Durante el período neonatal se sospechó síndrome de hipoventilación central congénita, lo que motivó la realización de estudio genético. Este detectó una mutación en heterocigosis en el gen PHOX2B. El paciente fue diagnosticado de síndrome de hipoventilación central congénita y manejado con ventilación mecánica domiciliaria invasiva mediante traqueostomía.
A los 5 meses de vida, el paciente reingresó con otro episodio caracterizado por signos y síntomas de obstrucción intestinal, con hallazgos en las pruebas de imagen que evidenciaban la presencia de vólvulo gástrico (más adelante se determinó que su causa fue la formación de adherencias tras la cirugía). El paciente fue sometido a gastropexia y, al sospecharse la EH, se obtuvieron muestras para biopsia durante la intervención. El estudio histológico reveló aganglionismo en el plexo mientérico e hiperplasia leve de neuronas colinérgicas en las muestras obtenidas del recto y el colon sigmoide. En este momento se programó la realización de un descenso transanal. Se realizó una primera intervención cuando el paciente tenía 11 meses, pero fue necesaria otra operación, ya que las muestras de biopsia del colon restante tras la primera resultaron ser agangliónicas. Al poco tiempo de la segunda operación se cerró la colostomía. Un mes después, el paciente presentó clínica compatible con nefrolitiasis en el lado izquierdo, tratada mediante litotricia. Las pruebas de imagen evidenciaron hidronefrosis, actualmente resuelta.
En meses posteriores, el paciente requirió múltiples dilataciones rectales por estenosis de la anastomosis, momento en que fue referido a nuestro centro. Presentó importante dermatitis perianal y perineal, que obligó a una ileostomía temporal. El curso de la enfermedad también se vio complicado por la malnutrición crónica (probablemente consecuencia de la asociación de EH, alergia a la proteína de la leche de vaca y síndrome de hipoventilación central congénita).
Actualmente el paciente tiene 9 años. Recibe irrigaciones rectales diarias, y ocasionalmente realiza deposiciones espontáneas. Requirió nutrición enteral nocturna de apoyo de forma temporal pero en los últimos años toma la mayor parte de su ingesta por vía oral. Se le diagnosticó de retraso cognitivo y del habla, con mucha mejoría desde que no requiere tantos ingresos hospitalarios.
DiscusiónEsta serie histórica de 1970 a 2013 muestra el progreso en el conocimiento de las mutaciones genéticas asociadas a la EH y sus implicaciones clínicas, que ha permitido mejorar el consejo prenatal y el uso de técnicas diagnósticas y profilácticas en los familiares de los afectados.
El gen RET, situado en el cromosoma 10q11, es el gen involucrado con mayor frecuencia en la EH, como se ha descrito en la literatura médica y observado en nuestro estudio. Es un receptor que regula la actividad de factores de crecimiento sobre las neuronas intestinales. Por lo tanto, es el primer gen que debería incluirse en el estudio genético tras diagnosticarse la EH. En nuestra serie, las mutaciones en este gen fueron el hallazgo más frecuente en el estudio genético
El gen PHOX2B, localizado en el cromosoma 4, codifica un factor de transcripción expresado en las células de la cresta neural que puede interferir en la expresión del gen RET. En la presente serie, 4 de los 160 pacientes (2,5%) tenían síndrome de Ondine. Solo se realizó estudio genético en 2 de ellos. En otro, solo se analizó el gen RET, en el que no se encontraron alteraciones. En el caso restante, se analizó el gen PHOX2B, en el que se detectó anormalidad. Por lo tanto, la prevalencia de mutaciones en el gen PHOX2B en la serie global fue del 0,63%, en comparación al 5% descrito en la literatura médica. Esto subraya la necesidad de incluir otros genes aparte del RET en el estudio genético cuando se sospecha un síndrome clínico o enfermedad asociada.
La importancia del estudio genético en la evaluación de la EH radica en sus implicaciones terapéuticas y pronósticas, no solo en caso de EH aislada, sino también en enfermedades asociadas. La implicación terapéutica y pronóstica más importante es que las mutaciones en ciertas regiones del gen RET se asocian al síndrome MEN2A o MEN2B24,25, asociados a un riesgo mayor de carcinoma medular de tiroides, hiperplasia de las glándulas paratiroides y feocromocitoma. En consecuencia, dado el mayor riesgo de desarrollar carcinoma medular de tiroides, los pacientes que tienen estas mutaciones han de ser sometidos a una tiroidectomía profiláctica26. Conocer la localización de la mutación es sumamente importante a efectos de tomar las medidas profilácticas apropiadas.
El estudio genético del gen RET en la EH beneficia no solo a los pacientes, sino también a sus familiares. Cuando se detecta una mutación, la extensión del estudio a los familiares de primer grado puede revelar la presencia de mutaciones previamente ignoradas en individuos asintomáticos que podrían ser susceptibles de tiroidectomía profiláctica y seguimiento especializado.
La descripción de las mutaciones asociadas a la EH, como las detectadas en el presente estudio, tiene una doble implicación. En primer lugar, una implicación clínica directa: es necesario conocer las mutaciones asociadas a la EH para que al detectarse en otros pacientes los clínicos sepan que son causantes de la enfermedad y no un hallazgo casual, y también conocer la localización exacta de la mutación en el gen, lo que permite determinar si esta se asocia a otras enfermedades o síndromes. En segundo lugar, un conocimiento mayor de las bases genéticas de la EH podría contribuir al esclarecimiento de la patogenia de la enfermedad en el futuro.
La principal limitación del estudio es el escaso número de pacientes sometidos a estudio genético en esta serie, lo que redujo la cantidad de datos genéticos relevantes documentados en los registros clínicos. De los 160 pacientes con diagnóstico confirmado de EH, tan solo 21 tenían resultados genéticos positivos registrados en su historial. No obstante, el estudio evidenció una conciencia creciente de la importancia del estudio genético a lo largo del tiempo, realizándose un número creciente de pruebas susceptibles de revisión y descripción en nuestro artículo, incluyendo aquellas que llevaron a la detección de 2 mutaciones nuevas. Todo ello apunta a una oportunidad de mejora mediante la inclusión rutinaria de pruebas genéticas en el protocolo de diagnóstico de pacientes con EH.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
