



La prematuridad con parálisis cerebral (PC) es frecuente en las consultas de neuropediatría. Las enfermedades neurodegenerativas como la ceroidolipofuscinosis neuronal (CLN) son infrecuentes y precisan alto grado de sospecha y una adecuada estrategia diagnóstica.
Las CLN son enfermedades lisosomales con herencia AR que afectan fundamentalmente a ojos y SNC. La clínica (pérdida de visión, demencia y epilepsia) es compartida por otros cuadros neurológicos progresivos.
El diagnóstico de las distintas formas de CLN clásicamente se basaba en la edad de inicio de la clínica y la anatomía patológica (AP): infantil, infantil tardía, juvenil y del adulto1,2. En la actualidad el diagnóstico de las CLN se basa en los hallazgos de la AP, los estudios enzimáticos y la genética molecular. Hay 8 formas con genes identificados: CLN10/CTSD, CLN1/PTT1, CLN2/TPP1, CLN3, CLN5, CLN6, CLN7, CLN8; CLN4 y CLN9 (todavía no identificado el gen)1,2. Todas las mutaciones están descritas en la NCL Mutation Database (http://www.ucl.ac.uk/ncl)3. En ocasiones hay dificultades para correlacionar fenotipo y genotipo.
Se presenta un caso de CLN infantil de inicio tardío y de difícil identificación por presentarse en niño con PC secundaria a prematuridad.
Segundo hijo de padres sanos no consanguíneos. Parto 28 semanas, peso 1.340 gramos, Apgar 4/9. Precisó ventilación mecánica 10 días, surfactante y drogas vasoactivas. Las ecografías neonatales mostraron hemorragia intraventricular grado ii. Desarrolló displasia broncopulmonar.
Durante su seguimiento, se objetivó PC tipo tetraparesia espástica iv según Gross Motor Function Classification System (GMFCS)4,5 con mayor afectación de hemicuerpo izquierdo.
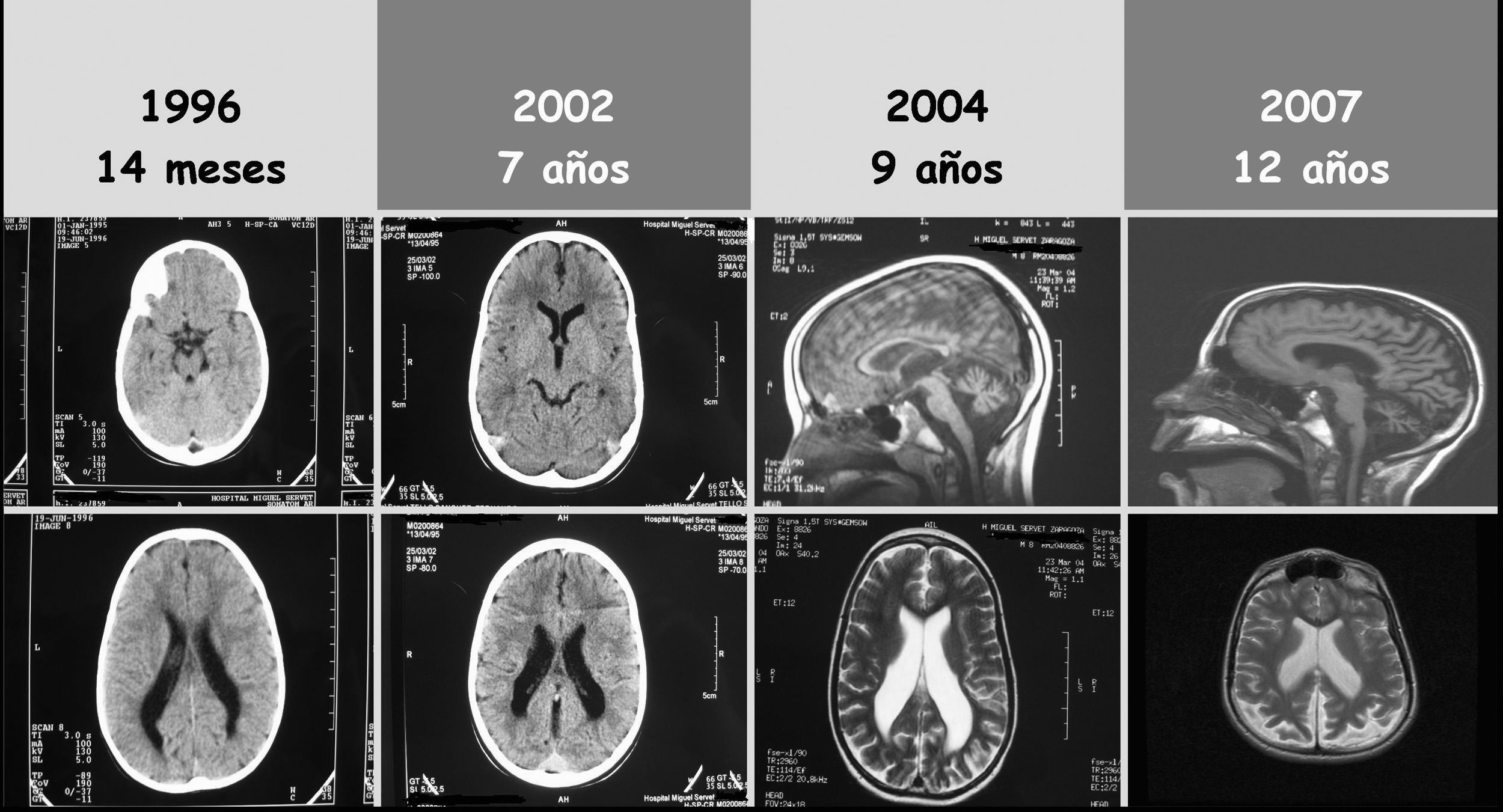
A los 14 meses, se realizó TAC craneal (fig. 1) compatible con leucomalacia periventricular (LMPV) del prematuro.
Mantenía buen contacto social, aceptable rendimiento escolar con adaptación curricular, y crecimiento del perímetro cefálico entorno P50.
A los 6 años se detectó hipoacusia neurosensorial que precisó prótesis auditivas.
A los 7 años comenzó con epilepsia, con varios tipos de crisis: parciales, ausencias, convulsiones y mioclonias. Hasta 3 días por semana y hasta 3 crisis diarias. Se realizó nueva TAC craneal (fig. 1) sin apreciarse cambios significativos. EEG mostraron anomalías focales y descargas de corta duración de punta-onda generalizadas. No hubo respuesta a distintos tratamientos: carbamacepina, oxcarbacepina, valproato, lamotrigina, topiramato, levetirazetam y clobazam. Desde el inicio de la epilepsia, se constató aparición progresiva de temblor, ataxia, babeo, disartria, empeoramiento de la visión y disminución del rendimiento escolar.
RM craneal, con 9 años (fig. 1), mostró LMPV conocida e inicio de atrofia cerebelosa y supratentorial de predominio posterior.
Con 12 años, el estudio oftalmológico mostró: importante afectación de la agudeza visual, nistagmus, fondo de ojo con papilas de aspecto atrófico y mácula con movilización pigmentaria. Los potenciales evocados visuales (PEV) fueron normales y el electrorretinograma no fue valorable. Perfil neurometabólico (hemograma, iones, gases, bioquímica, hormonas tiroideas, CK, amonio, láctico, 3-hidroxibutirato, homocisteína, cobre, ceruloplasmina, ácidos grasos de cadena muy larga, aminoácidos, sialotrasferrinas, ácidos orgánicos y test de toluidina): normal. Neurotransmisores en LCR sin alteraciones. EMG/ENG: sin hallazgos. RM cerebral, con 12 años, mostró aumento de atrofia cerebelosa y supratentorial de predominio posterior (fig. 1).
Se realizaron biopsias de músculo, piel, apéndice. El estudio de la cadena respiratoria no mostró alteraciones y no se apreciaron inclusiones lisosomales en microscopía electrónica (ME).
El análisis enzimático en fibroblastos (Dra. Chabás, IBC Barcelona) mostró deficiencia de tripeptidilpeptidasa i (TPP1) compatible con ceroidolipofuscinosis neuronal CLN2. El estudio genético (Dra. Milá, Hospital Clinic Barcelona), mediante secuenciación directa de la zona codificante del gen CLN2, mostró una mutación S381R (exón 10) no descrita que afecta al splicing además de cambiar el aminoácido, por lo que se considera causativa. Los cambios IVS7-10A>G y IVS8-18A>G son considerados potencialmente causativos aunque no descritos previamente. Está pendiente el estudio genético familiar para confirmar que los padres son portadores y que uno de estos cambios es realmente el responsable.
En la actualidad con 15 años, presenta: hipoacusia, ceguera, disfagia, afasia, temblor, ataxia, ausencia de control cefálico y de manipulación. Lleva tratamiento con valproato y clobazam sin control de las crisis.
No siempre es fácil correlacionar el fenotipo clínico con el genotipo en las CLN. Nuestro caso presenta dificultades añadidas. El tratarse de un niño con PC grave dificultó la valoración del empeoramiento clínico. La aparición de hipoacusia a los 6 años y epilepsia refractaria a los 7 años en niño con LMPV sin crisis previas, debieron ser las primeras pistas de enfermedad neurodegenerativa; sin embargo ambas son complicaciones frecuentes en el prematuro con LMPV. Por otra parte, la hipoacusia no es un signo típico en el inicio de CLN y tampoco el inicio de epilepsia a los 7 años, más tarde de lo habitual, en CLN26. Por último, no es del todo infrecuente la ausencia de inclusiones lisosomales en ME incluso en manos expertas, pues las inclusiones rara vez son extensas y de amplia distribución1,2.
Son necesarias estrategias adecuadas que permitan establecer diagnósticos difíciles. A destacar la importancia de la neuroimagen seriada cuando la evolución no es la esperada en un caso aparentemente cerrado. Es imprescindible contar con centros con experiencia en la ME, dada la dificultad de la misma. La existencia y continua aparición de numerosas mutaciones genéticas precisan una constante actualización de los centros de referencia en los estudios genéticos (http://www.ucl.ac.uk/ncl).
En la mayoría de casos de CLN en nuestro medio, dará el diagnóstico la determinación de la actividad enzimática en sangre (dry-spot)7 o fibroblastos (piel) de:
- •
Palmitoilproteína tioesterasa 1 (PPT1): CLN1 con gran variabilidad clínica de fenotipos infantil, infantil tardío, juvenil y del adulto.
- •
Tripeptidilpeptidasa I (TPP1): CLN2 con menos variabilidad clínica, fenotipos infantil tardía clásica y juvenil.
- •
Catepsina D (CTS): CLN10 forma congénita. Posteriormente se haría el estudio genético.
La forma CLN3 (juvenil o enfermedad de Batten) tiene presentación típica con ceguera progresiva, y como screening, podrían buscarse linfocitos vacuolados y luego realizar genética CLN3, más común 1kb deleción.
Si las determinaciones anteriores fueran negativas y persiste alto grado de sospecha de CLN, sería obligatoria la ME y si fuera positiva hacer genética orientada según ME de las formas menos habituales: CLN5 (forma finlandesa), CLN6, CLN7 (forma turca), CLN8 (forma turca y epilepsia del norte con retardo mental) y CLN4 gen todavía no identificado (forma del adulto o enfermedad de Kufs).
Queremos destacar las ventajas que a nuestro juicio aporta la biopsia de piel8, que permite: el estudio de la actividad enzimática (PTT1, TPP1, CTSD) y ME en las CLN; el estudio de otras enfermedades lisosomales y mitocondriales; y almacenar fibroblastos, tejido vivo, para futuros estudios en caso de no llegar a un diagnóstico.
