






La fibrosis quística (FQ) es la enfermedad autosómica recesiva grave más frecuente de la raza caucásica. El aumento tan importante de la supervivencia de los pacientes con FQ en los últimos años se debe en gran parte a la disponibilidad de nuevos tratamientos de la enfermedad pulmonar, en especial de las infecciones respiratorias.
Este protocolo revisa y actualiza los aspectos del diagnóstico y el manejo de la enfermedad respiratoria de la FQ. La prueba del sudor continúa siendo el estándar del diagnóstico de la FQ aunque, pese a realizarse de la forma adecuada, no siempre es concluyente.
Se debe controlar a los pacientes con FQ en unidades especializadas por un equipo multidisciplinario y experto, mediante protocolos específicos de seguimiento clínico, estudios por imagen y pruebas funcionales respiratorias y microbiológicas que se actualizan en esta revisión.
Se incluyen las recomendaciones sobre el tratamiento precoz y agresivo de la primoinfección y de la infección crónica por Pseudomonas aeruginosa, Staphylococcus aureus y otros microorganismos no habituales. Asimismo se describen los tratamientos de los diferentes aspectos de la enfermedad pulmonar y sus complicaciones y se revisan las indicaciones y el estado actual del trasplante pulmonar.
Este documento de consenso ha sido elaborado por los miembros del Grupo de Trabajo de Fibrosis Quística de la Sociedad Española de Neumología Pediátrica con el objetivo de actualizar el previo, publicado en esta Revista en 1999.
Cystic fibrosis (CF) is the most common severe recessive genetic disease in Caucasians. During the last years, new therapies and aggressive management of the lung disease have contributed significantly to the increased life expectancy in CF patients.
A review and update of CF diagnosis and management of lung disease are included.
The sweat chloride test (SCT) remains the gold standard for CF diagnosis and should be performed properly. However, in a few patients SCT results may not be conclusive to clarify the CF diagnosis.
Patients with CF should be followed up in specialist Units by an expert multidisciplinary expert applying standard clinical protocols and using lung function tests, and microbiological and imaging studies.
An overview with the recommendations for treatment of early onset and chronic infections due to Pseudomonas aeruginosa, Staphylococcus aureus and other uncommon pathogens is included. Furthermore, the management of other aspects of CF lung disease and complications is provided, as well as the indications for lung transplantation.
This document has been prepared by the members of the CF working group of the Spanish Paediatrics Pulmonary Society to provide an update to the earlier documents published in this Journal in 1999.
La fibrosis quística (FQ) es una enfermedad causada por mutaciones en el gen que codifica la proteína CFTR (cystic fibrosis transmembrane conductance regulator). Su funcionamiento defectuoso ocasiona una alteración del transporte de cloro y sodio en las células secretoras epiteliales, lo que da lugar a la aparición de manifestaciones clínicas multisistémicas, de las que las más relevantes son las del tracto respiratorio (afección pulmonar progresiva) y del sistema digestivo (insuficiencia pancreática y hepatopatía), sin olvidar otras como la deshidratación por pérdida de iones por el sudor o la infertilidad masculina por atresia o ausencia de los conductos deferentes. Debido a la complejidad de la enfermedad, se aconseja que los pacientes sean tratados en unidades especializadas con equipos multidisciplinarios que cuenten con profesionales entrenados en su diagnóstico y seguimiento, pues esta circunstancia mejora la calidad de vida y la supervivencia de los pacientes.
Este documento de consenso ha sido elaborado por miembros del Grupo de Trabajo de Fibrosis Quística de la Sociedad Española de Neumología Pediátrica con el objetivo de actualizar el previo, publicado en esta Revista en 1999.
Diagnóstico de la fibrosis quística a partir de la presentación clínicaLa prueba del sudor continúa siendo hoy la herramienta más útil para el diagnóstico. Los recientes Consensos de la European Cystic Fibrosis Society1 y el de la Cystic Fibrosis Foundation (CFF) de Estados Unidos2 lo han puesto de nuevo en el centro, al reconocer las limitaciones del conocimiento sobre la repercusión clínica real de muchas de las más de 1.500 mutaciones asociadas a la FQ. Por lo tanto, la calidad en su realización es esencial.
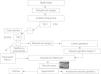
De acuerdo con el Consenso Europeo, se establece el diagnóstico de “fibrosis quística clásica” en presencia de al menos una característica fenotípica de FQ (enfermedad sinopulmonar crónica, alteraciones digestivas y nutricionales, síndromes de pérdida de sal, o ausencia bilateral de conductos deferentes), junto con una concentración de Cl en sudor ≥60mmol/l. En estos pacientes generalmente se detectan dos mutaciones causantes de enfermedad en el gen CFTR, pueden tener o no insuficiencia pancreática y su evolución clínica es variable. Se establece el diagnóstico de “fibrosis quística no clásica o atípica” si se halla al menos una de las características fenotípicas citadas y la prueba del sudor con resultado borderline (Cl 30–60mmol/l) junto con la detección de dos mutaciones y/o una diferencia de potencial nasal (DPN) alterado. Estos pacientes generalmente tienen suficiencia pancreática y enfermedad pulmonar más leve que los que sufren una FQ clásica, y la afección clínica puede ser de uno o varios órganos.
El Consenso de la CFF no hace la distinción entre FQ clásica y no clásica, pero establece un intervalo de normalidad para las concentraciones de Cl en sudor diferente según la edad. En lactantes, concentraciones<30mmol/l son normales y las de 30–59mmol/l, borderline. En pacientes de más edad, considera normales concentraciones<40mmol/l; borderline, las de 40–59mmol/l, e indicio de FQ, las >60mmol/l. En adultos señala que se necesitan más estudios sobre los límites de la normalidad de la concentración de Cl en sudor ya que, en un reciente estudio en 282 individuos sanos de 5 a 68 años, encontraron a 3 adultos (1%) con Cl ≥60mmol/l2.
La única prueba del sudor aceptable para la confirmación del diagnóstico es el test cuantitativo de iontoforesis con pilocarpina (QPIT)2, entendiéndose por tal el realizado por personal experto, con estimulación de la sudoración mediante iontoforesis con pilocarpina, recogida de la muestra mediante uno de los dos únicos procedimientos validados (papel de filtro o gasa prepesados, según la descripción originaria de Gibson y Cooke) o el método Macroduct, que utiliza un disco cóncavo y tubo espiral de plástico para la recogida del sudor y, por su sencillez, es el habitualmente realizado y cuyo procedimiento resumiremos. En ambos casos se debe analizar en el laboratorio la muestra, determinándose la concentración de Cl con un cloridómetro para micromuestras mediante coulombimetría (esto es, la titulación del cloruro de plata que se forma ante la exposición de un electrodo de plata a una solución que contiene cloro, lo que genera una diferencia de potencial) y, si es posible, también la de sodio mediante un fotómetro de llama; no es aceptable analizar únicamente in situ la conductividad eléctrica del sudor.
La muestra mínima de sudor para analizar debe ser de 75mg con el método de Gibson y Cooke y de 15μl con el Macroduct. Esta cantidad debe obtenerse en 30min de recogida, pues prolongarla para aumentar el tamaño de la muestra se asocia al riesgo de falsos negativos, por proceder de glándulas estimuladas subóptimamente. Tampoco es admisible combinar las muestras procedentes de dos recogidas para aumentar el tamaño. Sí se puede obtener dos muestras el mismo día, una de cada antebrazo.
Otros métodos utilizados en los laboratorios de bioquímica para determinar la concentración de cloro, como los colorimétricos o la potenciometría indirecta, aunque muy precisos en otros fluidos corporales, no están adecuadamente validados para los análisis de muestras de sudor, aunque afortunadamente hoy ya podemos disponer de cloridómetros adecuados para este fin fabricados en la Unión Europea.
De momento, no está validado para efectuar el diagnóstico de FQ, otro sistema (Nanoduct) que mide la conductividad eléctrica del sudor en muestras diminutas.
Protocolo del test del sudor mediante el sistema MacroductEstimulación (iontoforesis con pilocarpina) y recogida mediante el Macroduct Sweat Collection System (Wescor Inc., Logan, Utah Estados Unidos)3.
EstimulaciónSe lava cuidadosamente con agua destilada y alcohol la piel del antebrazo, y se procede a la estimulación siguiendo las instrucciones de los fabricantes. Se aplica una corriente de 1,5mA durante 5min con control automático de la intensidad y la duración de la estimulación. La corriente pasa entre los dos electrodos a los que se ha aplicado el reactivo de pilocarpina suministrado con el equipo.
RecogidaAl término de la estimulación se quitan los electrodos, se lava de nuevo la piel con agua destilada y se seca con toallitas de papel. Se coloca el sistema recolector (el disco cónico cóncavo invertido con agujero central conectado a tubo espiral de plástico), se tapa y se fija. Se permite que el sudor fluya durante 30min y rellene el tubo espiral. Pasado ese tiempo, se retira la fijación y se estima visualmente la cantidad de color recogida por el tramo de tubo que está teñido con el colorante marcador de color azul.
AnálisisEn primer lugar se recoge con aguja y jeringa el sudor del tubo espiral desenrollado, y se hace que fluya a la célula de conductividad de que está provisto el sistema (Sweat Check), la cual se calibra periódicamente según las instrucciones de los fabricantes. Se determina la conductividad, cuyo resultado se muestra digitalmente en pantalla. A continuación, se recupera con la jeringa la cantidad de sudor introducida en la célula y, junto con el resto del sudor, se transfiere a los recipientes facilitados por los fabricantes para el transporte de las muestras al laboratorio (siempre que el volumen sea ≥15μl). Las muestras se guardan en nevera hasta su traslado al laboratorio lo antes posible, y en todo caso antes de 48h, para la determinación de la concentración de cloro. Los protocolos de diagnóstico recomiendan determinar la concentración de Cl siempre que la conductividad sea ≥50mmol/l. La constatación en dos muestras de concentraciones de Cl en el sudor >60mmol/l es compatible con FQ clásica. El Consenso Europeo1 considera las concentraciones de Cl 30–60mmol/l como borderline, y motivo para realizar un estudio genético. No obstante, en casos con Cl borderline puede ser útil la medición de la relación Cl/Na. Raramente en controles es >1.
Se pueden dar falsos negativos de la prueba del sudor, en pacientes con edema. Se ha señalado la posibilidad de falsos positivos —generalmente basados en muy escaso número de observaciones— en una serie de entidades cuyas manifestaciones son muy diferentes de las de la FQ, y habitualmente no suponen problema diagnóstico alguno2. La gran mayoría de las causas de falsos negativos y positivos en el test del sudor son metodológicas. La contaminación de la muestra, especialmente en niños con piel atópica, por no seguir meticulosamente el protocolo de lavado, limpieza y secado de la piel antes y después de la iontoforesis, la utilización de otros métodos para la recogida del sudor sólo aceptables como cribado o el análisis únicamente de la conductividad eléctrica del sudor y/o de la concentración de sodio son frecuentes fuentes de error.
La figura 1 representa esquemáticamente el protocolo diagnóstico que propone el Consenso Europeo de 2006, corregido según las recomendaciones de la CFF, para el ajuste de los límites de la normalidad del Cl en sudor según la edad. Además de las observaciones anteriores sobre la variación con la edad de los límites de la concentración de Cl que son razonables para solicitar un estudio genético (panel de mutaciones), antes de ampliar este estudio a un rastreo completo de la región codificante y regiones intrónicas limítrofes del gen CFTR, hay que tener en cuenta la cobertura (porcentaje de alelos mutantes en los pacientes con FQ identificados) del kit genético comercial que se utliza en la población específica FQ a la que pertenece el paciente.
También hay que tener gran cautela antes de otorgar un valor decisivo a las determinaciones del DPN. La superposición de valores de esta determinación entre las poblaciones FQ y control es muy superior a la que se da con la prueba del sudor, y se necesita una gran experiencia con esta técnica.
Cribado neonatal de fibrosis quísticaDesde hace años se sabe que el diagnóstico precoz de la FQ mejora su pronóstico. La comprobación por Crosley en 1979 de que la tripsina estaba elevada en la sangre de cordón de los recién nacidos con FQ4 posibilitó la realización de un cribado inicial de esta enfermedad en el periodo neonatal. En 1983, la fundación estadounidense para la FQ recomendó la determinación de tripsina inmunorreactiva (TIR) utilizando una muestra de sangre del talón como método de cribado. Posteriormente se han desarrollado diferentes algoritmos que realizan una o dos determinaciones de TIR y, tras la identificación del gen, además se procede al estudio genético de las mutaciones de FQ más frecuentes en la población de la que procede el individuo.
En España el programa de cribado se lleva a cabo en la actualidad en siete comunidades: Cataluña, Castilla y León, Galicia, Extremadura, Baleares, Aragón-Rioja y Murcia. En la figura 2 se resume el programa de cribado iniciado en Cataluña hace 9 años, con el que se ha estudiado a más de 600.000 recién nacidos en esta comunidad. En este protocolo se realiza una doble determinación de TIR en papel secante y, en caso de que ambas sean positivas, se procede al estudio genético de las 32 mutaciones más frecuentes en la zona y una prueba del sudor. En caso de que no se identifiquen las dos mutaciones, se procede a la secuenciación completa del gen.
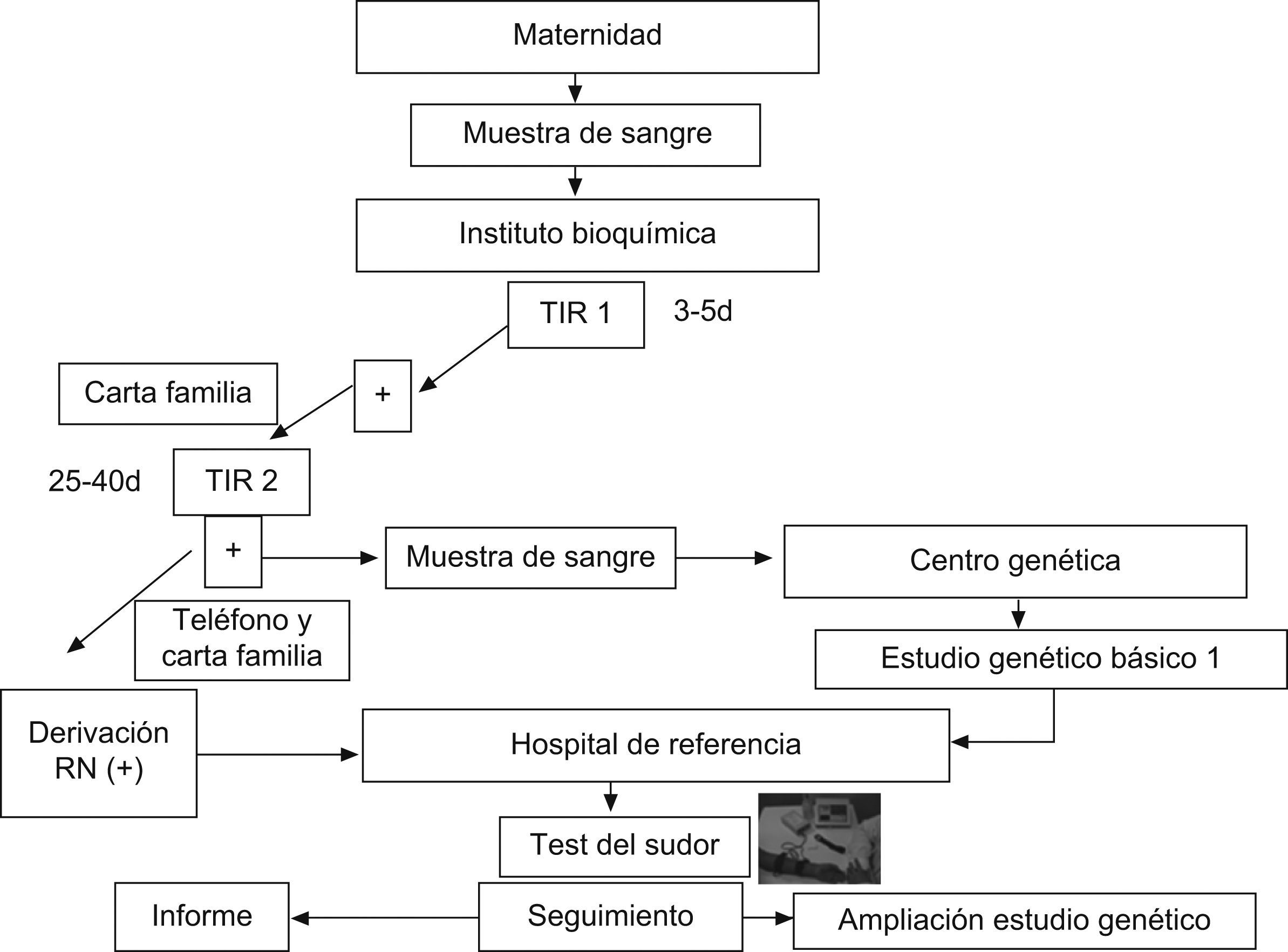
La aplicación de este programa ha permitido conocer la prevalencia de esta enfermedad en Cataluña, que se sitúa en 1/5.609 recién nacidos, y detectar síntomas, complicaciones graves y colonizaciones precoces por microorganismos que el tratamiento adecuado y temprano puede evitar o minimizar5,6. Sin embargo, la puesta en marcha de un programa de cribado también conlleva nuevos retos, como el diagnóstico de una enfermedad potencialmente grave en sujetos asintomáticos. Por ello, la información a los padres requiere una estrategia prudente y bien definida, especialmente en las formas atípicas o no clásicas de la enfermedad. En cualquier caso, dada la amplia variabilidad de la afección clínica, se recomienda individualizar las estrategias de seguimiento y tratamiento.
Evaluación y seguimientoEl seguimiento neumológico del paciente con fibrosis quística debe apoyarse en los siguientes puntos:
- 1.
Clínica. En cada visita se explorará al paciente y se registrará la presencia de síntomas respiratorios (tos, cambios en la cantidad o la coloración de la expectoración, tolerancia al esfuerzo, etc., así como el incremento ponderal o la presencia de problemas digestivos). En la exploración se registrarán las constantes vitales, el peso y la talla con sus correspondientes percentiles, y es aconsejable realizar mediciones antropométricas completas al menos anuales. Se explorará al paciente buscando anomalías en la auscultación pulmonar y signos de insuficiencia respiratoria: tórax abombado, cifosis, tiraje, cianosis, acropaquias. También existen varios sistemas de puntuación para medir de forma general la evolución de los pacientes, aunque ninguno de ellos refleja bien los sintomas respiratorios y tampoco detecta el inicio de las exacerbaciones. Entre todos ellos, el de Schwachman-Kulczycki es el más difundido7 (tabla 1).
Tabla 1.Puntuación de Schwachman-Kulczycki (modificada por Doershuk)
Puntos Actividad general Exploración física Nutrición Radiografía de tórax 25 Normal No tose Peso y talla >p-25 No enfisema Tolerancia al ejercicio normal Pulso y respiración normales Tono y masa muscular normales No aumento de trama Desarrollo motor normal No enfisema Grasa subcutánea normal No infiltrado o atelectasia Personalidad normal Auscultación normal Buen apetito Asistencia escolar normal No acropaquias Heces normales o casi Maduración sexual normal 20 Ligera limitación a la actividad vigorosa Tos débil ocasional Talla y peso >p-10 Enfisema mínimo Cansancio al final del día o tras ejercicio prolongado Carraspera. Pulso y respiración normales Rudeza respiratoria Normal tono y masa musculares Ligero aumento de la trama broncovascular Menos energético Roncus o espiración alargada ocasional y generalmente localizada Ligera disminución de grasa subcutánea No infiltrados Ocasionalmente irritable o pasivo No cargado de hombros Apetito normal No atelectasias Escolarización aceptable Acropaquias + Heces más frecuentes y ligeramente anormales Leve retraso de maduración sexual 15 Descansa durante el día Tos leve crónica no repetitiva al levantarse, después del ejercicio o con el llanto u ocasionalmente por el día. No tos nocturna Talla y peso >p-3 Enfisema moderado Se cansa tras el ejercicio Leve ↑ aumento de FC y FR Peso menor que talla ↑ Diámetro AP Regular asistencia al colegio ↑ Diámetro AP tórax Regular tono y masa musculares Pulmón más radiolucente Moderadamente inactivo Roncus o sibilancias ocasionales, estertores gruesos localizados Grasa subcutánea disminuida Diafragmas moderadamente deprimidos Ligero retraso motor Moderadamente cargado de hombros Falta de apetito Aumento de la trama Pasivo o irritable Acropaquias +/++ Distensión abdominal ligera Atelectasias localizadas o parcheadas Heces anormales, pero formadas Infiltración transitoria ocasional Retraso de maduración sexual 10 Actividad física y tolerancia al ejercicio limitadas Tos crónica frecuente, repetitiva, productiva y rara vez paroxística Talla y peso Enfisema marcado Disnea tras ejercicio Moderado ↑ de FC y FR Peso menor que talla ↑↑ Diámetro AP Moderado retraso motor Enfisema moderado-grave, tórax deformado, estertores, roncus o sibilancias generalmente presentes y a menudo generalizadas Escaso tono y masa musculares Diafragmas ↓↓ deprimidos Quisquilloso, irritable Cargado de hombros, cabeza adelantada Grasa subcutánea: ↓ marcada Silueta cardíaca pequeña Pobre escolarización Acropaquias ++/++++ Poco apetito Atelectasia generalizadas Profesor particular Heces poco formadas, grasas, voluminosas y malolientes A veces segmento o lobar Perezoso, apático Fallo de maduración sexual sin brote de crecimiento puberal Infiltrados persistentes Quistes localizados ↑ Marcado de la trama 5 Limitación importante de la actividad Tos grave, paroxística, frecuente y productiva, a menudo emetizante y hemoptoica. Tos nocturna Malnutrición y talla baja Cambios extensos Disnea y ortopnea Taquipnea y taquicardia No crece, a menudo pierde peso Enfisema grave Inactivo y confinado en cama o silla Tórax rígido, enfisema grave Débil, poca musculatura Atelectasia e infiltrado generalizados Retraso motor grave Estertores finos generalizados, roncus, sibilancias y espiración audible Ausencia de grasa subcutánea Quistes generalizados Apático o irritable Mala postura Heces voluminosas, frecuentes, malolientes y grasas Bronquiectasias, abscesos No puede ir al colegio Acropaquias +++/++++ Abdomen distendido Atelectasia lobar persistente A menudo cianosis A menudo prolapso rectal - 2.
Pruebas de imagen. Aunque en los niños más pequeños o en las formas más leves de la enfermedad la radiografía de tórax puede ser normal, se aconseja realizarla en cuanto se diagnostique al paciente. Posteriormente, y de forma progresiva, pueden ir apareciendo signos de atrapamiento aéreo, imágenes de consolidación (neumonía, atelectasia), bronquiectasias, lesiones quísticas y finalmente imágenes de fibrosis pulmonar. Es aconsejable utilizar un sistema de puntuación para evaluar las alteraciones radiológicas; los más utilizados son el de Crispin-Norman8, el Brasfield9 y el Northen CF Score10 que sólo precisa la proyección anteroposterior. La tomografía computarizada de alta resolución (TCAR) pone de manifiesto cambios radiológicos más precozmente que la radiografía de tórax, y además identifica con mayor precisión áreas de afección focal y es capaz de detectar bronquiectasias en ramificaciones bronquiales más finas. Por último, los cortes en espiración permiten identificar zonas de atrapamiento aéreo focal que indiquen afección de la pequeña vía aérea. También existen varios sistemas de puntuación (Nathanson, Santamaría, Brody, Helbich, Robinson), de los que quizá el de Bhalla11 sea el que obtiene la mejor correlación con la función pulmonar, especialmente en menores de 12 años. La TCAR es más sensible y específica que la radiografía de tórax y tiene una elevada correlación con la clínica, la función pulmonar y la propia radiografía de tórax. Entre sus inconvenientes están que se debe disponer de aparatos caros, la necesidad de emplear sedación en los niños pequeños no colaboradores y la mayor dosis de radiación. Esto puede evitarse utilizando dosis bajas (100kVp y 20-40mA) y realizando seis cortes en inspiración y cuatro en espiración. Aunque no hay acuerdo unánime sobre cuándo debe practicarse una TCAR en el seguimiento del paciente FQ, sí debería realizarse siempre que haya que tomar una decisión terapéutica o en el caso de complicaciones.
- 3.
Microbiología. La vigilancia microbiológica mediante el cultivo de esputo se realizará cada 1-2 meses y en las reagudizaciones respiratorias. En los pacientes incapaces de expectorar se recogerá una muestra mediante frotis faríngeo con tos inducida, aspirado nasofaríngeo o esputo inducido. Aunque el lavado broncoalveolar sería el método ideal, éste sólo se practicará en situaciones especiales. Se aconseja tratar de forma individualizada las muestras de estos pacientes en laboratorios de microbiología especializados para poder identificar microorganismos no habituales.
- 4.
Función pulmonar y gases sanguíneos. La afección respiratoria inicial se manifiesta en las vías aéreas periféricas con aumento del aire atrapado y disminución del FEF25–75%. Al progresar la enfermedad pulmonar también disminuye el FEV1 (volumen espirado en el primer segundo). Este parámetro se correlaciona con la progresión y la mortalidad de la enfermedad, por lo que en cuanto el paciente colabore debe realizarse espirometría en cada visita. Conviene realizar también una prueba con broncodilatador, para incluirlo o no en el tratamiento según la respuesta obtenida. La pletismografía se realizará opcionalmente en caso de que se disponga de ella. A medida que avanza la enfermedad se produce una disminución de la presión arterial de oxígeno y un aumento de la presión arterial de anhídrido carbónico, que en la actualidad se pueden monitorizar de forma no invasiva mediante pulsioxímetros y capnógrafos. Sería aconsejable realizar pulsioximetría nocturna a todos los pacientes con FEV1≤50% o con saturación de oxígeno en reposo entre el 93 y el 94%12.
Desde los primeros meses de la vida, algunos pacientes sufren colonización e inflamación crónica endobronquial. En la fase inicial es característica la presencia de Haemophilus influenzae y Staphylococcus aureus. Posteriormente casi todos los pacientes presentan colonización por Pseudomonas aeruginosa, que se asocia a un deterioro progresivo e irreversible de la función pulmonar; la infección endobronquial crónica por P. aeruginosa es la causa más importante de morbilidad y mortalidad de los pacientes con FQ. El tratamiento antibiótico dirigido contra este patógeno se ha convertido en la piedra angular para controlar la progresión de la enfermedad. La monitorización del esputo es importante para identificar el microorganismo y determinar a qué antibióticos es sensible13. Éstos se pueden administrar por vía oral, inhalada o intravenosa, dependiendo de la situación clínica del paciente, el patógeno aislado y su antibiograma14.
Los pacientes con FQ presentan una farmacocinética diferente, especialmente con los aminoglucósidos y los betalactámicos, que los individuos normales. El volumen de distribución de estos fármacos por kilo de peso está aumentado, la eliminación por la vía renal es mayor y, como consecuencia, su vida media está disminuida. Por lo tanto, resulta razonable utilizar dosis más altas y monitorizar las concentraciones alcanzadas en sangre con el fin de minimizar su riesgo potencial de ototoxicidad y nefrotoxicidad.
Tratamiento de Staphylococcus aureusEn la fase inicial de la enfermedad, el paciente suele ser colonizado por S. aureus con o sin clínica de exacerbación respiratoria. Aunque el papel del tratamiento profiláctico con betalactámicos ha mejorado notablemente el pronóstico de estos pacientes, hay controversia respecto al resultado del tratamiento largo e indiscriminado con estos antibióticos, ya que podría favorecer la aparición de P. aeruginosa y acelerar el progreso de la enfermedad. No hay consenso alguno sobre el tratamiento de S. aureus y la mayoría de los centros tratan sólo las exacerbaciones agudas según el antibiograma (tabla 2).
Antibióticos empleados en la afección respiratoria en la fibrosis quística
| Antibiótico | Dosis | Vía de administración | Frecuencia (h) | |
| Penicilinas | Amoxicilina | 50–100mg/kg/día | Oral | 8 |
| Amoxicilina-ácido clavulánico | 40–80mg/kg/día | Oral | 8 | |
| 100mg/kg/día | i.v. | 8 | ||
| Cloxacilina | 100mg/kg/día | i.v. | 6 | |
| 50mg/kg/día | Oral | 6 | ||
| Piperacilina/ticarcilina | 300mg/kg/día | i.v. | 6 | |
| Cefalosporinas | Cefuroxima | 30mg/kg/día | Oral | 12 |
| Ceftazidima | 200mg/kg/día | i.v. | 8 | |
| Cefepima | 150 mg/kg/día | i.v. | 8 | |
| Otros betalactámicos | Aztreonam | 150mg/kg/día | i.v. | 6 |
| Imipenem | 50–100mg/kg/día | i.v. | 6 | |
| Meropenem | 60–120mg/kg/día | i.v. | 6–8 | |
| Aminoglucósidos | Amikacina | 20–30mg/kg/día | i.v. | 24 |
| Gentamicina | 6–15mg/kg/día | i.v. | 24 | |
| Tobramicina | 5–10mg/kg/día | i.v. | 24 | |
| Tobramicina | 300mg | Inhalado | 12 | |
| Otros antibióticos | Ciprofloxacino | 20–40mg/kg/día | Oral | 12 |
| 10–20mg/kg/día | i.v. | 12 | ||
| Colimicina | 1–2×106U | Inhalado | 12 | |
| Trimetroprima-sulfametoxazol | 6mg/kg/día | Oral | 12 | |
| 10mg/kg/día | i.v. | 12 | ||
| Vancomicina | 60mg/kg/día | i.v. | 12 |
En las etapas iniciales también se encuentra H. influenzae pero, a diferencia de lo que ocurre con S. aureus, disminuye el porcentaje de aislamiento durante los primeros años y en raras ocasiones se aísla de forma crónica. Como en el caso anterior, la mayoría de los centros lo tratan sólo en las exacerbaciones y según antibiograma.
Tratamiento de P. aeruginosaDe acuerdo con los patrones microbiológicos de la colonización infección pulmonar por P. aeruginosa, hemos de diferenciar los conceptos de colonización inicial con o sin signos de infección, colonización esporádica o intermitente y colonización crónica en fase estable o en fase de exacerbación.
En 2005 se publicó el primer consenso español sobre el tratamiento antibiótico contra la colonización por P. aeruginosa15, donde quedan reflejadas las siguientes indicaciones:
- •
Si se detecta la aparición de P. aeruginosa, tratamiento precoz y agresivo con antibióticos inhalados y antibióticos por vía oral o intravenosos.
- •
Tratamiento de mantenimiento con antibióticos inhalados, con o sin antibióticos orales o intravenosos.
- •
Tratamiento de las exacerbaciones pulmonares con antibióticos orales o intravenosos.
La eliminación de P. aeruginosa de las secreciones bronquiales de estos pacientes es prácticamente imposible, pues tras su instauración en la vía aérea produce una matriz (alginato) que, además de permitirle formar colonias, actúa como una barrera que impide a los antibióticos alcanzar una concentración adecuada. Cuando se aísla P. aeruginosa por primera vez, se debe iniciar de inmediato un tratamiento combinado con antibióticos inhalados y orales o intravenosos. El consenso español recomienda efectuar tratamiento de la primocolonización por P. aeruginosa sin signos de infección con ciprofloxacino oral a la dosis de 15–20mg/kg/12h durante 21 días y un antibiótico por vía inhalatoria, ya sea tobramicina en solución para inhalación o colimicina. Al cabo de 1 mes, se repite el cultivo. Si el cultivo resulta positivo, se administra un nuevo ciclo de ciprofloxacino oral de 21 o 30 días, manteniendo la antibioterapia inhalatoria. Si pasados 1 o 2 meses más el cultivo todavía persiste positivo, se pautará un ciclo de antibióticos intravenosos de 14 días de duración. Si se sigue cultivando el microorganismo, se considerará situación de colonización crónica. Si por el contrario el cultivo se ha negativizado cuando transcurran 6-12 meses, se puede suspender el antibiótico inhalado.
En la actualidad se dispone de tobramicina inhalada, específica para nebulización, en dosis de 300mg dos veces al día, administrada en ciclos intermitentes de 28 días de duración. También existe una amplia y prolongada experiencia con la colimicina inhalada, a la dosis de 1–2×106U/12h en régimen continuo, con resultados similares a los obtenidos con los aminoglucósidos y escaso desarrollo de resistencias16. Las dosis utilizadas se detallan en la tabla 2.
Los antibióticos inhalados se deben administrar con compresores de alto flujo, ≥6–8l/min, y es preferible utilizar un nebulizador que dispense la medicación sólo en la fase inspiratoria, para aprovechar mejor la dosis administrada y disminuir la contaminación ambiental. En la actualidad se dispone de aparatos electrónicos de malla vibradora como el e-Flow® y el INeb®. Con estos sistemas se logra mayor eficacia y más comodidad para el paciente. Con el e-Flow® se puede administrar tanto colimicina como tobramicina, mientras que con el INeb®, sólo colimicina. Éste presenta la novedad de incorporar el sistema AAD® (Adaptative Aerosol Delivery), que analiza el ritmo de respiración y sólo administra el fármaco cuando el paciente inspira y cuando realiza la técnica inhalatoria de manera correcta, lo que permite disminuir la dosis a la mitad.
En el tratamiento oral se utiliza el ciprofloxacino. Los posibles efectos secundarios que éste puede tener en el cartílago de crecimiento limitaron inicialmente su uso en la edad pediátrica. Sin embargo, se han realizado estudios ecográficos y de resonancia magnética en pacientes con FQ que recibieron ciprofloxacino y no se han evidenciado efectos adversos, aunque la experiencia en niños menores de 5 años es escasa. En algunos pacientes se puede desarrollar fotosensibilidad, que es controlable con el uso de pantallas solares. Las dosis utilizadas se detallan en la tabla 2.
Tratamiento de mantenimientoEn la situación de colonización crónica en fase estable, el paciente presenta siempre cultivos positivos o cuando durante un periodo de 6 meses presenta por lo menos tres cultivos positivos en muestras separadas entre sí más de 1 mes. El tratamiento de mantenimiento tiene como objetivos prevenir la infección o colonización crónica por P. aeruginosa, disminuir el número y la gravedad de las exacerbaciones pulmonares y enlentecer el círculo infección-inflamación que conduce al daño pulmonar irreversible. En algunas unidades se utilizan las denominadas pautas programadas de tratamiento antibiótico intravenoso cada 3-4 meses, incluso asumiendo que la situación clínica se halla estabilizada. Cada ciclo de tratamiento dura unos 14-21 días y combina generalmente un betalactámico y un aminoglucósido. La combinación más utilizada posiblemente sea la de ceftazidima a la dosis de 50–70mg/kg/8h o cefepima 50mg/kg/8h más tobramicina 10mg/kg/24 o amikacina 20–30mg/kg/24h. Cada vez con mayor frecuencia, el paciente se aplica estos ciclos de tratamiento intravenoso en su propio domicilio.
Otra modalidad de tratamiento de supresión crónica consiste en la terapia inhalatoria antimicrobiana diaria, que dura tanto como persista la colonización bacteriana. En los pacientes con afección pulmonar de base moderadamente grave, puede agregarse al tratamiento inhalado persistente un ciclo de 3-4 semanas de ciprofloxacino oral cada 3-4 meses. En los pacientes con afección grave, se recomienda realizar estos ciclos con antibioterapia intravenosa.
Tratamiento de las exacerbaciones pulmonaresEs característico de los pacientes con FQ que presenten exacerbaciones de la infección pulmonar (tabla 3). El tratamiento antibiótico que emplear puede ser por vía oral si la exacerbación es leve o moderada o por vía intravenosa si la exacerbación es moderada o grave. El régimen del tratamiento antibiótico puede ser hospitalario o domiciliario.
Exacerbación respiratoria
| Aumento de la tos |
| Aumento de la expectoración |
| Cambios de las características del esputo |
| Aumento de la disnea |
| Disminución de la tolerancia al ejercicio |
| Disminución del apetito |
| Aumento de la frecuencia respiratoria |
| Cambios en la auscultación pulmonar |
| Empeoramiento de la función pulmonar |
| Fiebre y leucocitosis |
| Pérdida de peso |
| Infiltrados radiológicos nuevos |
La terapia combinada de un betalactámico, como la ceftazidima, y un aminoglucósido es de elección para la mayoría de los centros. Las dosis utilizadas se detallan en la tabla 2. Últimamente, se ha preconizado el tratamiento con aminoglucósidos administrados una vez al día, con el objeto de lograr los mismos resultados terapéuticos y disminuir los efectos secundarios. En los casos de exacerbaciones por microorganismos multirresistentes, se trata según antibiograma. La duración del tratamiento oscila habitualmente entre 2 y 3 semanas, pero se puede prolongar en casos especiales.
Tratamiento intravenoso domiciliarioEsta modalidad se utiliza cada vez con mayor frecuencia, ya que reduce el número de ingresos hospitalarios y el gasto sanitario y proporciona al paciente mejor calidad de vida, al permitirle proseguir con la mayor parte de sus actividades. Para que un paciente sea apto para recibir un tratamiento intravenoso domiciliario, debe cumplir una serie de requisitos, como presentar una exacerbación moderada, tener una función renal normal, no requerir otro tipo de soporte terapéutico, no presentar reacciones adversas a los medicamentos y tener una vía de acceso venoso segura. La administración de la primera dosis siempre se debe realizar en el hospital, y después debe seguir controles semanales.
Infección por microorganismos poco habitualesEn los últimos años varias especies gramnegativas nuevas, intrínsicamente resistentes, como Burkholderia cepacia complex, Stenotrophomonas maltophilia, Achromobacter xylosoxidans, etc., se han hecho más comunes en los pacientes con FQ17. Tampoco es raro encontrar Aspergillus spp., Candida spp. y micobacterias atípicas. Todo ello podría deberse al incremento de la expectativa de vida, el uso frecuente de antibióticos y de medios más selectivos que permiten el aislamiento de nuevas especies bacterianas.
Burkholderia cepacia. Abarca un grupo de especies (B. cepacia complex) compuesto de diferentes “genomovares” o microorganismos genéticamente distintos, de los que se han identificado nueve hasta el momento. Los genomovares II y III representan el 85% de los aislados de B. cepacia complex en los pacientes con FQ, el 50% de ellos B. cenocepacia. El genomovar III es el que habitualmente causa las infecciones más graves y está ligado a epidemias y mal pronóstico en el trasplante pulmonar, de ahí que algunos grupos consideren su presencia como una contraindicación. B. cepacia se aísla hasta en el 7% de los pacientes con FQ. De ellos, un 20% contrae el “síndrome cepacia”, caracterizado por un rápido deterioro de la función pulmonar, frecuentemente acompañado de bacteriemia, que suele evolucionar hacia insuficiencia respiratoria. Recientes análisis multivariables han identificado su detección como un indicador independiente de mal pronóstico17. Suele aislarse junto a S. maltophilia y/o P. aeruginosa; el pronóstico cuando ambos están presentes suele ser peor que cuando sólo se cultiva P. aeruginosa. B. cepacia complex presenta un alto perfil de multirresistencias (>70%), que incluye cefalosporinas y aminoglucósidos. Es resistente a penicilinas (con sensibilidad variable a su asociación con los inhibidores de las betalactamasas), cefalosporinas de primera, segunda y tercera generación, pero no por completo a minociclina, meropenem, ceftazidima, cefepima, aztreonam y aminoglucósidos. B. cepacia es transmisible tanto dentro como fuera del hospital, por lo que se recomienda aislar a los sujetos colonizados o infectados para evitar la infección cruzada de otros pacientes con FQ.
Stenotrophomonas maltophilia. Se detecta hasta en el 15% de los pacientes con FQ. Aunque su significado clínico no está del todo claro18, se sabe que por sus propiedades inmunoestimuladoras e inductoras de la producción de factor de necrosis tumoral (TNF) alfa dan lugar a una significativa inflamación bronquial. Su aislamiento es más frecuente en pacientes de mayor edad, generalmente mujeres, con peor función pulmonar y mayor número de exacerbaciones, consultas, hospitalizaciones, ciclos de antibióticos intravenosos y terapia antibiótica a largo plazo. No se ha relacionado con hospitalizaciones recientes o tener hermanos colonizados por este microorganismo. Es intrínsecamente resistente a la mayoría de los antimicrobianos, pero son buenas opciones terapéuticas las nuevas fluoroquinolonas (moxifloxacino y levofloxacino) y el cotrimoxazol, que se considera de elección, aunque se recomienda combinarlo con otros fármacos como colimicina, ticarcilina-ácido clavulánico o doxiciclina.
Achromobacter (Alcaligenes) xylosoxidans. Se detecta hasta en el 18% de los cultivos a una media de edad de 20 años. Su significado clínico tampoco está claro, dado el bajo número de pacientes que presentan colonización crónica (alrededor del 5%). Se han documentado casos de deterioro clínico con exacerbaciones agudas, pero suelen producirse en pacientes también colonizados por P. aeruginosa. Se ignora cuál es su vía de infección, aunque no parece que el contacto persona a persona sea el principal mecanismo transmisor. Los factores de riesgo de colonización son la utilización previa de antibióticos, las hospitalizaciones frecuentes y, en alguna serie, la presencia de Aspergillus fumigatus en el esputo. Suele aislarse en pacientes con una función pulmonar más deteriorada, que no suele empeorar tras la colonización, excepto en los que presentan un rápido incremento de anticuerpos precipitantes específicos. En cambio, suelen precisar más ciclos de antibióticos intravenosos19. Más del 37% de las cepas son multirresistentes a colimicina, aminoglucósidos, penicilinas y cefalosporinas de primera y segunda generación. La resistencia a imipenem y meropenem puede llegar hasta el 40%, mientras que las nuevas fluoroquinolonas, minociclina y doxiciclina, son algo más activas.
S. aureus resistente a meticilina (SARM). Es un problema creciente en pacientes con FQ. Su presencia oscila entre el 2% (Hospital Brompton de Londres) y el 14,6%, según el último registro anual de la fundación estadounidense de FQ. En España se detecta en un 2,2%. Los pacientes que precisan ingresos hospitalarios frecuentes, más ciclos de antibióticos y los que están infectados crónicamente por Aspergillus fumigatus tienen mayor riesgo de contraer la infección. Aunque se ha asociado con deterioro respiratorio20, no está clara su repercusión en el curso de la enfermedad; sin embargo, crea problemas de control epidemiológico hospitalario y puede contraindicar el trasplante de pulmón. En el hospital se debe separar a estos pacientes del resto y, pese a que la transmisión entre pacientes con FQ no es frecuente, se aconseja evitar el contacto íntimo. Existen diversos grupos de antibióticos con actividad contra SARM. De ellos, los más utilizados en las infecciones agudas graves han sido la vancomicina y la teicoplanina, aunque actualmente linezolid supera en penetración pulmonar a los anteriores. Además, hay otros antibióticos de administración oral con cierta actividad anti-SARM, como rifampicina, fosfomicina, trimetoprima-sulfametoxazol, tetraciclinas, ácido fusídico, etc., pero con rápido desarrollo de resistencias si se administran como monoterapia. En el caso de pacientes con FQ, la dificultad para diferenciar la colonización de la auténtica infección hace que siga siendo discutible la necesidad de administrar tratamiento antibiótico.
Micobacterias no tuberculosas. Su prevalencia oscila entre el 4 y el 24%, pero las series que incluyen niños son escasas o utilizan técnicas de identificación o cultivos inadecuados. Los pacientes suelen ser de más edad y frecuentemente están colonizados por S. aureus pero no por P. aeruginosa. Las especies más comunes son Mycobacterium avium complex y M. abscessus. Su significación patógena vendría ligada a la existencia de múltiples cultivos positivos, la falta de respuesta al tratamiento antibacteriano convencional y la observación en la TCAR de nódulos pulmonares periféricos y lesiones granulomatosas, confirmadas mediante biopsia. En estos casos se recomienda iniciar tratamiento específico, que ha se demostrado que logra la mejoría clínica y la negativización de los cultivos. En el caso de M. avium, la combinación con rifampicina, claritromicina y etambutol debe mantenerse 12 meses tras la negativización de los cultivos. M. abscessus es particularmente resistente a la terapia y se utiliza imipenem intravenoso o cefoxitima más amikacina durante 1 mes, seguidos de claritromicina oral más etambutol durante al menos 12 meses tras la negativización21.
Hongos. La colonización/infección fúngica, sobre todo por hongos levaduriformes, no es infrecuente en estos pacientes, pero se desconoce su papel en el curso de la enfermedad. Los hongos detectados más frecuentemente son Candida albicans y A. fumigatus, y su presencia se ve favorecida por las bronquiectasias. Candida spp. y, en concreto, C. albicans se aíslan en un 50-75% de los casos; se lo considera un comensal inofensivo, pero se desconoce su implicación en el deterioro de la función pulmonar. Aspergillus spp. y, sobre todo, A. fumigatus aparecen en más del 25% de los pacientes con FQ, que también presentan títulos de anticuerpos específicos más altos que la población general. Su detección no se relaciona con la infección por P. aeruginosa. No suelen causar enfermedad invasiva, aunque ésta puede aparecer en los enfermos trasplantados. Sin embargo, en los que sufren deterioro clínico tras su detección sin respuesta a la terapia antibacteriana, debería considerarse el tratamiento antifúngico22. La aspergilosis broncopulmonar alérgica (ABPA) se produce por sensibilización a alérgenos de A. fumigatus presentes en el ambiente y se tratará de ella más adelante. Otros hongos filamentosos aislados con relativa frecuencia del tracto respiratorio del paciente con FQ son: Scedosporium apiospermum (hasta el 9% de casos y con probable acción patógena, ya que puede desencadenar una respuesta inflamatoria), Wangiella dermatitidis y Penicillium emersonii.
Tratamientos que actúan sobre el aclaramiento mucociliarDNasa recombinanteEl esputo purulento de los pacientes con FQ contiene grandes cantidades de ADN procedente de la destrucción de neutrófilos y bacterias. La DNasa actúa destruyéndolo, con lo que reduce la viscosidad de las secreciones y facilita su eliminación. Se ha visto que su empleo mejora la función pulmonar y disminuye las exacerbaciones, la inflamación bronquial y las imágenes de atrapamiento aéreo, aunque el significado clínico de estos hallazgos es dudoso.
Se recomienda en mayores de 6 años, empleando una ampolla de 2,5mg sin diluir una vez al día, utilizando un compresor de alto flujo para su nebulización. Dado que la respuesta al tratamiento es variable, se debe valorar su continuación tras su aplicación durante unos meses23.
Suero salino hipertónico inhaladoSe ha propuesto como tratamiento para aumentar la hidratación de la vía aérea y conseguir un mayor aclaramiento mucociliar. Consigue una disminución del número de exacerbaciones y una cierta mejoría de la función pulmonar24.
Se utiliza la concentración del 7% y volumen de 5–10ml dos veces al día. Debe de aplicarse con precaución en pacientes con hiperreactividad bronquial. Algunos autores aconsejan su empleo en pacientes mayores de 6 años, individualizando su tolerancia y su eficacia.
N-acetilcisteínaReduce la viscosidad del esputo al romper los puentes disulfuro y restaura la concentración de glutatión en los polimorfonucleares circulantes y en la vía aérea. Sin embargo, por el momento no hay pruebas suficientes para aconsejar su empleo en pacientes con FQ por vía oral o inhalada.
BroncodilatadoresUn número significativo de pacientes con FQ responde a los broncodilatadores, aunque la respuesta es variable y más evidente durante las exacerbaciones. Se pueden usar betaadrenérgicos, que añaden cierto efecto antiinflamatorio y antiadherencia bacteriana, o anticolinérgicos (más efectivos en adultos que en niños).
Se pueden emplear, a las dosis habituales, en pacientes que muestren mejoría del FEV1 tras su inhalación, y luego realizar controles periódicos, pues la respuesta no permanece invariable.
AntiinflamatoriosCorticoidesEl tratamiento a largo plazo con corticoides orales mostró beneficios iniciales en la función pulmonar que no se mantuvieron a largo plazo; unido a sus efectos secundarios, ello desaconseja su empleo prolongado25. En situaciones puntuales se podrían utilizar a dosis de 1mg/kg/día durante periodos cortos. El empleo de corticoides inhalados a largo plazo no ha resultado beneficioso26, por lo que su uso debe limitarse a los pacientes con FQ y clínica de asma. Probablemente sea necesario emplear dosis altas y valorar los dispositivos de inhalación, debido a la previsible disminución del depósito pulmonar por las secreciones espesas o cambios en el comportamiento físico de las partículas.
Otros tratamientosEl tratamiento a largo plazo con altas dosis de ibuprofeno se ha demostrado que ralentiza el declinar de la función pulmonar, sobre todo en los pacientes jóvenes27. Sin embargo, su administración a largo plazo puede elevar el riesgo de hemorragia gastrointestinal y requiere controles farmacocinéticos, por lo que su empleo no se ha generalizado. Los macrólidos, en especial la azitromicina, tienen actividad inmunomoduladora, y en enfermos colonizados crónicamente por P. aeruginosa disminuyen el número de exacerbaciones pulmonares y consiguen discreta mejoría de la función pulmonar. Se administra azitromicina 500mg tres veces a la semana en pacientes de más de 40kg y dosis de 250mg si su peso es<40kg28.
Rehabilitación respiratoriaLa rehabilitación comprende la fisioterapia respiratoria, el ejercicio gradual e individualizado, la terapia postural dirigida a la prevención de las deformidades y la educación sanitaria encargada de clarificar todos los aspectos relacionados con la enfermedad. Se debe iniciar en el mismo momento del diagnóstico, con un adecuado entrenamiento de los padres y el paciente.
Existen diferentes técnicas de fisioterapia respiratoria: técnica convencional de percusión y drenaje postural, técnica de ciclo activo, drenaje autógeno, presión positiva espiratoria (PEP) o mediante utilización de diversos dispositivos (Flutter o Cornet) y compresión torácica mediante chaleco o ventilador intrapulmonar percusivo.
No haya claras ventajas de una sobre otra y el método elegido ha de tener en cuenta la edad del paciente, su disposición y su autonomía. Se aconseja realizar dos sesiones diarias, y aumentar su frecuencia y su duración en las reagudizaciones. También se recomienda la práctica de ejercicio aeróbico reglado y programado, al menos 30min tres veces por semana.
ComplicacionesNeumotóraxAparece en el 5–8% de los pacientes con FQ, especialmente en aquellos con afección pulmonar moderada-grave, y la frecuencia llega a ser del 20% de los mayores de 18 años. La causa más frecuente es la rotura de ampollas subpleurales, aunque también podría deberse a los incrementos de volumen y presión alveolar causados por los tapones de moco.
Los neumotórax de tamaño pequeño (<20% del volumen del hemitórax afectado) y asintomáticos pueden tratarse con observación hospitalaria, reposo y oxigenoterapia. Si es sintomático, se aplica el mismo tratamiento que a los de gran tamaño. En este caso (>20% del volumen), la reexpansión puede conseguirse mediante un tubo torácico fino con sello de agua. En todo neumotórax que recidiva, se ha de considerar un tratamiento más agresivo, como la pleurodesis quirúrgica o química, que actualmente no contraindica la realización de trasplante pulmonar29.
HemoptisisSegún la cuantía de la hemorragia, se considerará esputo hemoptoico o hemoptisis leve (<30ml/día), hemoptisis moderada (30–150ml/día), hemoptisis grave (> 150ml/día) o hemoptisis masiva, que para el CFF Patient Registry se define como >240ml/día o que requiera transfusión. La hemoptisis amenazante lo será por el riesgo vital que representa (disnea, signos de hipovolemia) y en función del volumen perdido (>600ml/día), la velocidad de la hemorragia y la capacidad funcional cardiorrespiratoria de base. En este caso el paciente debe ser trasladado a la UCI y a los demás se los mantendrá en observación durante 6–8h.
Protocolo de actuaciónSangrado ocasional autolimitado o con persistencia de mínima expectoración hemoptoica: se realiza radiografía de tórax, hemograma, perfil hepático, estudio de coagulación y gasometría, y se deja al paciente en observación 6–8h.
Sangrado moderado o grave: se debe hospitalizar al paciente, con reposo en cama en decúbito homolateral, para cuantificar el volumen de la hemoptisis y monitorizar al paciente, asegurar la permeabilidad de la vía aérea y disponer de una vía venosa y de reserva de sangre. El tratamiento incluye antitusígenos, antibióticos intravenosos y oxigenoterapia continua.
Hemoptisis masivas: ingreso en UCI, donde pueda realizarse intubación selectiva.
La broncoscopia practicada durante la hemoptisis activa no la agrava y permite realizar varias técnicas, en general con buenos resultados pero de utilidad transitoria: lavados con suero fisiológico frío y adrenalina al 1/10.000 y bloqueo de la luz bronquial. La arteriografía bronquial mediante cateterización arterial es diagnóstica y terapéutica, pues permite localizar y embolizar los vasos sangrantes. Se puede valorar la resección quirúrgica del foco si está localizado, es accesible o cuando otras medidas transitorias son ineficaces.
Aspergilosis broncopulmonar alérgicaLa aspergilosis broncopulmonar alérgica (ABPA) se caracteriza por una respuesta inmunitaria contra antígenos de A. fumigatus que coloniza el árbol bronquial y se manifiesta con sibilancias, disnea, infiltrados pulmonares, bronquiectasias centrales y fibrosis30. En los pacientes con FQ su prevalencia oscila entre el 2 y el 8%, según los criterios diagnósticos utilizados. Aunque puede presentarse en menores de 6 años, es significativamente más frecuente entre los 10 y los 20 años.
La ABPA se puede manifestar como un empeoramiento agudo de su función respiratoria, con incremento de la tos y de la expectoración, que frecuentemente muestra tapones mucosos marrones. Puede haber sibilancias, fiebre y aparición de nuevos infiltrados pulmonares en la radiografía de tórax.
En cuanto al diagnóstico, en 2003 se publicó un consenso auspiciado por la Cystic Fibrosis Foundation30, en la que se propusieron nuevos criterios. Diagnóstico de ABPA clásica:
- •
Deterioro clínico agudo o subagudo no atribuible a otra etiología.
- •
IgE >1.000U/ml, a menos que el paciente esté recibiendo corticoides sistémicos.
- •
Pruebas cutáneas positivas a A. fumigatus o hallazgo de IgE específica contra A. fumigatus.
- •
Positividad de precipitinas o IgE específica.
- •
Alteraciones radiológicas nuevas que no mejoran tras tratamiento con antibióticos y fisioterapia.
Criterios diagnósticos mínimos:
- •
Deterioro clínico agudo o subagudo no atribuible a otra etiología.
- •
IgE sérica >500U/ml. Si se sospecha ABPA y la IgE está entre 200 y 500U/ml, se recomienda nueva determinación en 1-3 meses.
- •
Pruebas cutáneas o IgE específica contra A. fumigatus positivas.
- •
Una de las siguientes: a) precipitina o IgG sérica contra A. fumigatus, o b) alteraciones radiológicas nuevas o recientes que no desaparecen con antibióticos y fisioterapia.
Como método de criba de ABPA, el consenso propone:
- •
Mantener un alto nivel de sospecha en pacientes mayores de 6 años y hacer una determinación anual de IgE. Si ésta fuera >500U/ml, deberían realizarse pruebas cutáneas y determinación de IgE especifica contra A. fumigatus. Si el resultado fuera positivo, hay que considerar el diagnóstico de ABPA basado en criterios diagnósticos mínimos.
- •
Si la IgE total estuviera entre 200 y 500U/ml, se recomienda repetir la determinación, y si hay sospecha clínica de ABPA, se deben realizar las pruebas mencionadas anteriormente.
En cuanto a la determinación de anticuerpos IgE contra alérgenos recombinantes mayores de A. fumigatus, aunque no hay unanimidad entre los autores y el número de publicaciones es escaso, parece que los pacientes con ABPA presentan aumento de IgE contra rAspf2, rAspf4 y rAspf6.
El tratamiento se inicia con prednisona o equivalente (0,5–2mg/kg/día, máximo 60mg/día) durante 1-2 semanas, para ir disminuyéndola durante 2-3 meses, según resultados de IgE total, radiología, espirometría y síntomas respiratorios. La administración conjunta de antifúngicos (itraconazol, voriconazol) mejora la respuesta al tratamiento corticoideo31 y permite disminuir su administración, con el beneficio de minimizar sus efectos adversos.
Oxigenoterapia y ventilación no invasiva en la FQEl deterioro progresivo de la función pulmonar en el paciente con FQ se produce por la obstrucción inflamatoria de la vía aérea que causa bronquiectasias, acumulación de moco y destrucción del parénquima pulmonar. A medida que van disminuyendo el FEV1 y la FVC, los músculos respiratorios se ven obligados a trabajar más para mantener un adecuado intercambio de gases32. Cuando la enfermedad está muy avanzada o en las exacerbaciones respiratorias graves, el paciente sufre taquipnea con respiración superficial como mecanismo compensador de la sobrecarga de los músculos respiratorios para vencer las importantes resistencias pulmonares. A pesar de ello, se produce una hipoventilación alveolar progresiva que conduce a hipoxemia y finalmente también a hipercapnia. La oxigenoterapia crónica domiciliaria compensa la hipoxemia, pero no corrige la hipercapnia, incluso puede empeorarla si se incrementa rápidamente el aporte de oxígeno, en cuyo caso el paciente sufre signos y síntomas de hipercapnia severa.
Cuando el paciente está hipoxémico sólo durante el sueño, se puede indicar oxigenoterapia nocturna para mantener una SpO2 normal, siempre y cuando no retenga CO2. Mediante un concentrador de O2 u oxígeno líquido, con un flujo en la cánula nasal de 1–3l/min, le proporcionaremos el aporte necesario de O2 para corregir la hipoxemia sin generar hipercapnia. Es conveniente realizar durante una noche un registro con pulsioximetría y capnografía para ajustar el tratamiento. Si el paciente tiene hipoxia o disnea diurna, se debe indicar oxigenoterapia continua (o tantas horas como sea capaz de tolerar) para evitar o retrasar la situación de cor pulmonale.
La ventilación no invasiva (VNI) se ha demostrado eficaz en pacientes con insuficiencia respiratoria crónica, fundamentalmente en enfermos con trastornos restrictivos pulmonares. También se ha utilizado en sesiones de fisioterapia respiratoria para evitar la fatiga muscular respiratoria y facilitar la eliminación del esputo. Una revisión Cochrane33 acerca de la aplicación de VNI para la FQ concluye que podría ser útil como complemento de la fisioterapia respiratoria. En pacientes con enfermedad moderada o grave que precisen oxigenoterapia durante el sueño, utilizada como complemento podría mejorar el intercambio gaseoso en mayor grado que el tratamiento con oxígeno solamente.
Trasplante de pulmónLa FQ es la indicación más frecuente de trasplante pulmonar en niños. Representa en el registro internacional el 55% de los trasplantes en el grupo de edad entre 6 y 11 años y el 69% en los niños de 12-17 años34.
Hay evidencia firme de que el trasplante pulmonar aumenta la supervivencia de los niños afectos de FQ con enfermedad pulmonar grave35. Sin embargo, un estudio reciente ha cuestionado este hecho concluyendo que el trasplante pulmonar no alarga la vida a los niños menores de 18 años afectos de FQ36. Las conclusiones de ese trabajo han recibido muchas críticas, ya que el análisis practicado se basa en datos no totalmente adecuados de los pacientes y se realiza una interpretación equivocada37,38.
En cualquier caso, mientras el trasplante no consiga prolongar la vida indefinidamente, es necesario predecir el momento evolutivo en que un paciente afecto de FQ obtendrá el máximo beneficio del trasplante. Esta es una de las decisiones más complicadas a que se enfrentan los equipos de trasplante: valorar el momento adecuado para incluir a un niño con FQ en lista de espera para trasplante. Se considera recomendable hacerlo si pese a estar recibiendo el máximo tratamiento médico su esperanza de vida es<2 años y tiene una mala calidad de vida que puede mejorar con el trasplante37. Sin embargo, la progresión de la enfermedad es muy variable y en la práctica la decisión para indicar un trasplante pulmonar considera las siguientes variables: FEV1<30% respecto al teórico, deterioro rápido de la función pulmonar, desnutrición resistente a una intervención nutricional agresiva, cor pulmonale, hipoxemia (pO2<55mmHg) y/o hipercapnia (pCO2 >50mmHg) e incremento progresivo de la resistencia antimicrobiana de los patógenos pulmonares. Los pacientes más jóvenes y las mujeres son los que tienen más riesgo. También se ha relacionado con mayor mortalidad el descenso del pico de consumo de oxígeno y una reserva respiratoria baja en el umbral anaeróbico en la prueba de esfuerzo cardiopulmonar39.
En resumen, las indicaciones para remitir a un paciente para que sea evaluado en una unidad con programa de trasplante pulmonar serían: hospitalizaciones frecuentes para antibioterapia intravenosa, limitaciones importantes para ir al colegio, descenso rápido o fluctuaciones marcadas de la función pulmonar, como FEV1<30%, microorganismos con resistencia antimicrobiana en aumento, hipoxemia e hipercapnia39.
Muchos centros consideran la colonización por Burkholderia cepacia una contraindicación absoluta para la realización del trasplante, dado que se ha descrito un riesgo de muerte precoz tras el trasplante del 50%. El análisis molecular ha permitido definir que este mal pronóstico se relaciona fundamentalmente con la infección por B. cepacia genomovar III, mientras que es mejor la evolución con otros subtipos de esta bacteria. El aislamiento de P. aeruginosa multirresistente o A. fumigatus no constituye una contraindicación para el trasplante. La colonización por hongos multirresistentes como Scedosporium prolificans puede ensombrecer el pronóstico. La realización previa de una pleurodesis no suele ser contraindicación. La necesidad de ventilación mecánica no invasiva previa al trasplante tampoco lo es, pero el pronóstico del trasplante es peor en los niños que requieren ventilación mecánica con intubación traqueal por una agudización respiratoria.
Tras el trasplante pulmonar se produce una mejoría muy importante en la calidad de vida de los niños trasplantados. El 90% de los niños a los 3 años del trasplante y el 80% a los 7 años no tienen ninguna limitación en su actividad. La mayoría de los receptores de un trasplante pulmonar tienen un desarrollo y una función cognitiva normales, aunque un pequeño porcentaje presenta dificultades psicológicas y una calidad de vida afectada38.
En España las cifras de supervivencia al trasplante pediátrico están entre el 62 y el 70% a los 5 años y en el 62% a los 8 años.
Hasta que no se consiga una cura para los niños enfermos de FQ con insuficiencia respiratoria terminal, el trasplante pulmonar es una opción terapéutica efectiva.
