




La prevalencia de las hemoglobinopatías en nuestro medio ha aumentado como consecuencia de los flujos migratorios.
La talasemia maior cursa con anemia hemolítica crónica y necesidad de transfusiones regulares desde el año de vida. La enfermedad drepanocítica cursa con anemia, vasculopatía y daño orgánico progresivo. En ambas, la esperanza de vida está disminuida. El trasplante alogénico de progenitores hematopoyéticos es una opción de curación para estos pacientes.
PacientesDiecisiete pacientes recibieron un trasplante alogénico de progenitores hematopoyéticos: 14 afectados de talasemia maior y 3 de enfermedad drepanocítica.
ResultadosLos donantes fueron en los pacientes con talasemia maior 9 hermanos HLA-idénticos, 2 progenitores con una diferencia antigénica HLA y 3 donantes no emparentados, y en aquellos con enfermedad drepanocítica, 3 hermanos HLA-idénticos. La fuente fue la médula ósea en todos, excepto uno.
La mediana de edad al trasplante de progenitores hematopoyéticos fue de 6 años (intervalo: 1-16) en los niños con talasemia maior y doce años (intervalo: 8-15) en los niños con enfermedad drepanocítica.
Se confirmó injerto medular en todos los pacientes. Dos con talasemia maior presentaron fallo de injerto secundario, precisando nuevamente soporte transfusional. Trece pacientes presentaron quimerismo completo y 2 quimerismo mixto, todos con normalización de la cifra de hemoglobina y sin requerimiento de transfusiones. Los pacientes con enfermedad drepanocítica no presentaron más episodios vasooclusivos y la funciones pulmonar y cerebral en aquellos pacientes que presentaban afectación en el momento del trasplante se estabilizaron. Tres pacientes con talasemia maior desarrollaron enfermedad injerto contra huésped crónica y 5, hipogonadismo hipogonadotropo.
ConclusionesNuestra experiencia confirma que el trasplante alogénico de progenitores hematopoyéticos de hermano HLA idéntico es una buena opción en el tratamiento de la talasemia maior y la enfermedad drepanocítica. En el caso de la talasemia maior, el trasplante de donante no emparentado es una opción terapéutica en centros especializados, ya que, a pesar de los buenos resultados presentados, la morbimortalidad de este procedimiento puede ser elevada, frente a la alternativa de un tratamiento médico no curativo pero con expectativas de supervivencia prolongada. En el caso de la enfermedad drepanocítica, el trasplante de donante no emparentado todavía está en fases preliminares de investigación.
The prevalence of hemoglobinopathies in Spain is increasing as a result of immigration.
Thalassemia major presents with chronic hemolytic anemia that requires regular red blood cell transfusions within the first year of life. Patients with sickle cell disease suffer from chronic anemia, vasculopathy and progressive damage in almost any organ. There is decreased life expectancy in both conditions. Allogeneic hematopoietic stem cell transplantation represents the only potentially curative option.
PatientsSeventeen patients (fourteen thalassemia major, and three sickle cell disease) underwent allogeneic hematopoietic stem cell transplantations.
ResultsIn the thalassemia group, nine donors were HLA-geno-identical siblings, two were partially matched related donors (one HLA allele mismatch), and three unrelated donors. All three patients with sickle cell disease were transplanted from HLA-geno-identical siblings. The source of stem cells was bone marrow in sixteen cases.
Median patient age at transplant was six years (range: 1–16) in the thalassemia group, and twelve years (range: 8–15) in the sickle cell disease group.
The graft was successful in all patients. Secondary graft rejection was observed in two thalassemia patients rendering them dependent on blood transfusions. Complete chimerism was observed in thirteen patients and, although mixed chimerism occurred in two, with all of them showing normal hemoglobin levels after transplantation and not requiring further transfusion support. Patients affected by sickle cell disease did not present with new vaso-occlusive crises, and stabilization of pulmonary and neurological function was observed. Chronic graft-versus-host disease was detected in three patients affected by thalassemia, and hypogonadotrophic hypogonadism in five patients.
ConclusionsWe conclude that for thalassemia major and sickle cell disease, allogenic hematopoietic stem cell transplantation from HLA-geno-identical siblings offers a high probability of complication-free survival. Despite good results, morbidity and mortality associated with transplantation from unrelated donors is a risk that might be considered, in contrast to a non-curative medical treatment that offers a long term survival. For thalassemia major groups it could be an option, but not for sickle cell disease, which is still in the investigational phase.
Las hemoglobinopatías son un conjunto de enfermedades hereditarias caracterizadas por una síntesis, estructura y/o función anormal de la molécula de la hemoglobina. La Organización Mundial de la Salud estima que anualmente 330.000 recién nacidos presentan desórdenes hemoglobínicos, siendo los más graves la talasemia maior (TM) y la enfermedad drepanocítica (ED)1. Su prevalencia es extremadamente alta en países del Sudeste asiático, Oriente Medio, África, Caribe y la cuenca mediterránea, siendo excepcional en nuestro medio2, donde como consecuencia de los flujos migratorios, ambas afecciones han aumentado su prevalencia.
La TM es una enfermedad autosómica recesiva caracterizada por la producción desequilibrada de las cadenas de globina, generando una eritropoyesis inefectiva con hemólisis extravascular aumentada. Los niños afectados presentan anemia hemolítica crónica desde el primer año de vida, dependiente de transfusiones de hematíes. La esperanza de vida de estos pacientes, a pesar de recibir transfusiones periódicas, era de apenas 10 años. Con la introducción de la quelación de hierro, esta situación cambió, permitiendo una esperanza de vida muy superior. Sin embargo, aquellos pacientes con una quelación no óptima desarrollan complicaciones secundarias a la sobrecarga férrica que pueden llevar a un fallo multiorgánico progresivo. El trasplante alogénico de progenitores hematopoyéticos (alo-TPH) ha sido hasta el momento el único tratamiento curativo1,2. Recientemente, se ha descrito el primer paciente tratado con terapia génica3.
La ED es una enfermedad autosómica recesiva caracterizada por la presencia de hemoglobina falciforme (Hb S), que confiere al hematíe una forma de hoz característica, ocasionando vasooclusión y hemólisis. Los niños afectados presentan anemia hemolítica crónica, lesión isquémica aguda, lesión isquémica crónica, asplenia funcional secundaria a múltiples infartos y riesgo de enfermedad neumocócica invasiva. La lesión crónica de los órganos afectados (especialmente pulmón y SNC) conlleva una esperanza de vida de alrededor de 35 años1. Las medidas profilácticas, como la vacunación frente a bacterias encapsuladas y el tratamiento con penicilina, así como el uso de hidroxiurea en pacientes seleccionados, han permitido mejorar la esperanza de vida de estos pacientes, pero resultan insuficientes para frenar el carácter progresivo de la enfermedad1. El alo-TPH es el único tratamiento curativo4.
Presentamos los resultados del alo-TPH en niños con TM y ED en nuestro centro.
Pacientes y métodosEstudio retrospectivo descriptivo de pacientes pediátricos afectados de TM o ED sometidos a alo-TPH en nuestro centro, previo consentimiento informado de padres o tutores, entre noviembre de 1989 y septiembre del 2010.
El diagnóstico de la enfermedad de base se realizó mediante electroforesis de hemoglobina, excepto en un caso, en que el diagnóstico fue prenatal a través de técnicas de biología molecular.
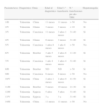
Los niños afectados de TM, previamente al alo-TPH, fueron asignados a un grupo de riesgo según los criterios de la clasificación de Pesaro en New England Journal of Medicine en 19905. Se realizó alo-TPH a todos aquellos que tenían un hermano HLA-idéntico. En 2 pacientes con un cumplimiento irregular del tratamiento médico y, consecuentemente, con una elevada sobrecarga férrica, sin hermano HLA idéntico, se consideró como tratamiento de elección el trasplante de un progenitor con una única diferencia antigénica HLA. A partir de 2008, se inició el programa de alo-TPH de donante no emparentado (DNE) y los pacientes clase 1 y 2 según los criterios de Pesaro que no disponían de donante familiar HLA compatible pero con un DNE idéntico en 10/10 locus fueron incluidos en él. La fuente de progenitores hematopoyéticos de elección fue la médula ósea. El régimen de acondicionamiento varió según el grupo de riesgo y el tipo de donante. Los pacientes con alo-TPH de donante familiar clase 1 y 2 recibieron busulfán (BU) vo 14mg/kg o equivalente iv, y ciclofosfamida (CF) iv 200mg/kg. Los alo-TPH de donante familiar clase 3 recibieron BU vo 14mg/kg o equivalente iv, CF 160mg/kg y globulina antitimocítica (GAT) 7,5mg/kg. Un paciente clase 3 con focos de hematopoyesis extramedular hepáticos y esplénicos recibió radioterapia nodal. Desde el año 2008, a los regímenes descritos se añadió tiotepa 10mg/kg. Los pacientes sometidos a alo-TPH de DNE recibieron BU iv en dosis según peso, CF 200mg/kg, GAT 7,5mg/kg y tiotepa 10mg/kg (tabla 1). Aquellos pacientes no clasificables según los criterios de Pesaro, recibieron el régimen de acondicionamiento correspondiente a clase 1 y 2, excepto uno de los casos, correspondiente a una paciente cuyo diagnóstico fue tardío y que en el momento del diagnóstico presentaba unas manifestaciones clínicas avanzadas, que recibió el régimen descrito para clase 3. La profilaxis de enfermedad injerto contra huésped (EICH) se realizó con ciclosporina y metotrexato. Como profilaxis antiinfecciosa, se procedió a aislamiento en cámaras de flujo laminar y profilaxis antimicrobiana según pauta habitual de la unidad (tabla 1).
Características del trasplante
| Paciente/sexo | Diagnóstico | Estadio enf. previo a alo-TPH | Edad al alo-TPH | Tipo alo-TPH alogénico HLA-idéntico | Fuente | Acondicionamiento |
| 1/H | Talasemia | – | 6 años | DNE | mo | Bu + Cy + GAT + TT |
| 2/V | Talasemia | – | 1 año 1 meses | Madre | mo | Bu + Cy |
| 3/V | Talasemia | Pesaro 2 | 13 años 7 meses | Hermano | mo | Bu + Cy |
| 4/V | Talasemia | Pesaro 1 | 2 años 10 meses | Padre | sp | Bu + Cy |
| 5/V | Talasemia | Pesaro 3 | 14 años 6 meses | Hermano | mo | Bu + Cy + GAT + RT |
| 6/V | Talasemia | – | 4 años 1 meses | Hermano | mo | Bu + Cy + TT |
| 7/V | Talasemia | – | 4 años 3 meses | Hermana | mo | Bu + Cy |
| 8/H | Talasemia | – | 4 años 1 meses | Hermano | mo | Bu + Cy + GAT |
| 9/H | Talasemia | Pesaro 3 | 16 años 5 meses | Hermana | mo + scu | Bu + Cy + GAT |
| 10/V | Talasemia | – | 4 años 7 meses | DNE | mo | Bu + Cy + GAT + TT |
| 11/H | Talasemia | Pesaro 3 | 8 años 10 meses | Hermana | mo | Bu + Cy + GAT |
| 12/H | Talasemia | Pesaro 3 | 9 años 6 meses | Hermana | mo | Bu + Cy + GAT + MEL |
| 13/V | Talasemia | Pesaro 3 | 8 años 1 meses | Hermana | mo | Bu + Cy + GAT + MEL |
| 14/H | Talasemia | – | 5 años 9 meses | DNE | mo | Bu + Cy + GAT + TT |
| 15/V | Drepanocitosis | Síndrome torácico agudo | 12 años 3 meses | Hermano | mo | Bu + Cy + GAT |
| 16/V | Drepanocitosis | Nefropatía | 8 años 2 meses | Hermana | mo | Bu + Cy + GAT |
| 17/V | Drepanocitosis | Vasculopatía SNC | 15 años 0 meses | Hermana | mo | Bu + Cy + GAT |
| Profilaxis EICH | Injerto | >0,5 × 10e9/l neutrófilos | >20 × 10e9/l plaquetas | > 1% hematíes | Quimerismo | Pérdida implante |
| CsA + MTX | Sí | 21 | 33 | 30 | 100% D | No |
| CsA + MTX | Sí | 22 | 23 | 18 | 100% D | No |
| CsA + MTX | Sí | 30 | 42 | 20 | 83% D, 17% R | No |
| CsA + MTX | Sí | 15 | 15 | 15 | 100% D | No |
| CsA + MTX | Sí | 19 | 14 | 24 | 100% D | No |
| CsA + MTX | Sí | 26 | 32 | 15 | 100% D | No |
| CsA + MTX | Sí | 23 | 31 | 35 | 100% D | No |
| CsA + MTX | Sí | 25 | 33 | 40 | 100% D | Sí |
| CsA + MTX | Sí | 17 | 20 | 20 | 100% D | No |
| CsA + MTX | Sí | 19 | 25 | 19 | 100% D | No |
| CsA + MTX | Sí | 21 | 31 | 20 | 50% D, 50% R | Sí |
| CsA + MTX | Sí | 16 | 79 | 26 | 100% D | No |
| CsA + MTX | Sí | 15 | 69 | 68 | 100% D | No |
| CsA + MTX | Sí | 23 | 39 | 43 | 100% D | No |
| CsA + MTX | Sí | 21 | 28 | 19 | 100% D | No |
| CsA + MTX | Sí | 15 | 29 | 12 | 100% D | No |
| CsA + MTX | Sí | 28 | 26 | 28 | 100% D | No |
Bu: busulfán; CsA: ciclosporina; Cy: ciclofosfamida; D: donante; GAT: gammaglobulina antitimocítica; MEL: melfalán; mo: médula ósea; MTX: metotrexato; R: receptor; sp: sangre periférica; TT: tiotepa.
Dosificación de los quimioterápicos utilizados:
Bu: dosis oral: 14mg/kg o dosis intravenosa equivalente (14 dosis, dosis ajustada según peso: < 9kg: 1mg/kg/dosis; 9-16kg: 1,2mg/kg/dosis; 16-23kg: 1,1mg/kg/dosis; 23-34kg: 0,95mg/kg/dosis; > 34kg: 0,8mg/kg/dosis), Cy iv: 160 o 200mg/kg, según grupo de riesgo y donante GAT: 7,5mg/kg, TT: 10mg/kg, MEL: 140mg/m2.
En los pacientes afectados de ED, la indicación de alo-TPH se estableció según los criterios de elegilibilidad de Walters en New England Journal of Medicine en 19966. El donante fue en todos los casos un hermano HLA-idéntico. La fuente de progenitores hematopoyéticos fue la médula ósea. El régimen de acondicionamiento fue BU vo 16mg/kg o equivalente iv, CF 200mg/kg y GAT 7,5mg/kg (tabla 1). La profilaxis de la EICH se realizó con ciclosporina y metotrexato. Como profilaxis antiinfecciosa se procedió a aislamiento en cámaras de flujo laminar y profilaxis antimicrobiana según pauta habitual de la unidad. Se realizó profilaxis anticomicial con fenitoína en todos los pacientes hasta la supresión de ciclosporina6,7.
En la evolución postrasplante se tuvieron en cuenta el prendimiento del trasplante, el desarrollo de EICH, el quimerismo, la electroforesis de hemoglobina postrasplante, la desaparición de necesidades transfusionales y la aparición de complicaciones tardías.
ResultadosEntre noviembre de 1989 y septiembre del 2010, 14 pacientes afectados de TM y 3 de ED recibieron alo-TPH en nuestro centro.
Características de los pacientesTalasemia maiorEntre los pacientes afectados de TM, 8 eran niños y 6, niñas. La mediana de edad al diagnóstico fue de 11 meses (intervalo: 0 meses-5 años). El diagnóstico más precoz fue prenatal, a raíz del antecedente de un hermano afectado. El diagnóstico más tardío fue en una paciente originaria de Egipto tras su llegada a nuestro país, no habiendo sido previamente estudiada ni transfundida. La mediana de edad a la que recibieron la primera transfusión fue de 16 meses (intervalo: 3 meses-5 años). En 8 de los 14 niños, la primera transfusión se realizó en el momento del diagnóstico. Desde el diagnóstico hasta el alo-TPH pasó una mediana de 5 años (intervalo: 10 meses-16 años). Antes del alo-TPH, 12 pacientes habían recibido más de 20 transfusiones, con sobrecarga férrica secundaria en todos ellos, siendo la mediana del valor de la ferritina en el momento previo al alo-TPH de 1,844 ng/ml (intervalo: 698–8,158 ng/ml). En 12 de los 13 pacientes en los que se estudió, se demostró, bien mediante biopsia hepática bien mediante RMN hepática, la existencia de depósitos de hierro patológicos. Doce recibieron tratamiento quelante de hierro, con estabilización posterior de los niveles de ferritina. En 2 casos, se realizó una esplenectomía, por aumento progresivo de las necesidades transfusionales secundario a hiperesplenismo.
En función de la clasificación de Pesaro5, un paciente fue catalogado como clase 1, uno como clase 2 y 5 como clase 3. Siete pacientes no fueron clasificables según la clasificación de Pesaro al no haberse realizado biopsia hepática.
Las características de los pacientes se detallan en la tabla 2.
Características de los pacientes
| Paciente/sexo | Diagnóstico | Etnia | Edad al diagnóstico | Edad 1.ª transfusión | N.° transfusiones pre alo-TPH | Hepatomegalia |
| 1/H | Talasemia | China | 11 meses | 11 meses | > 50 | No |
| 2/V | Talasemia | Gitana | 3 meses | 3 meses | < 10 | Sí |
| 3/V | Talasemia | Caucásica | 11 meses | 3 años 1 meses | 31-40 | Sí |
| 4/V | Talasemia | Gitana | 0 meses | 3 meses | 31-40 | Sí |
| 5/V | Talasemia | Caucásica | 1 años 8 meses | 1 año 8 meses | > 50 | No |
| 6/V | Talasemia | Bereber | 3 año 5 meses | 2 años 8 meses | 11-20 | Sí |
| 7/V | Talasemia | Caucásica | 1 año 4 meses | 1 años 4 meses | 31-40 | Sí |
| 8/H | Talasemia | Bereber | ND | ND | > 50 | Sí |
| 9/H | Talasemia | Caucásica | 6 meses | 6 meses | > 50 | No |
| 10/V | Talasemia | China | 2 años 1 meses | 2 años 9 meses | 41-50 | Sí |
| 11/H | Talasemia | Bereber | 5 meses | 10 meses | 21-30 | Sí |
| 12/H | Talasemia | Egipcia | 5 años | 5 años | 31-40 | Sí |
| 13/V | Talasemia | Egipcia | 3 a | 3 a | 31-40 | Sí |
| 14/H | Talasemia | China | 11 meses | 11 meses | > 50 | No |
| Ferritina, ng/ml | Biopsia hepática patológica | RMN hepática patológica | Fibrosis portal | Esplenectomía | Quelación | Pesaro |
| 1,162 | – | Sí | – | No | Deferoxamina + deferiprona | – |
| 1,382 | - | Sí | - | No | - | - |
| 1,569 | Grado I | No | No | No | Deferoxamina | 2 |
| 1,610 | Grado II | Sí | No | Sí | Deferoxamina | 1 |
| 1,720 | Grado IV | – | Sí | No | Deferoxamina | 3 |
| 1968 | – | Sí | – | Sí | Deferasirox | – |
| 2017 | – | – | – | No | Deferoxamina | – |
| 2080 | – | Sí | – | No | Deferasirox | – |
| 2,710 | Grado III | Sí | Sí | No | Deferoxamina | 3 |
| 3,600 | – | Sí | – | No | Deferasirox | – |
| 4,554 | Grado IV | Sí | Sí | No | Deferoxamina | 3 |
| 698 | Grado I | Sí | Sí | No | – | 3 |
| 8,158 | Grado II | Sí | Sí | No | Deferoxamina | 3 |
| 921 | – | No | – | No | Deferoxamina + deferiprona | – |
| Paciente/sexo | Diagnóstico | Etnia | Edad al diagnóstico | Edad 1.ª transfusión | N.° transfusiones pre alo-TPH | Hepatomegalia |
| 15/V | Drepanocitosis | Mora | 2 años | 6 años | < 10 | No |
| 16/V | Drepanocitosis | Subsahariana | 1 año 7 meses | 1 año 6 meses | < 10 | Sí |
| 17/V | Drepanocitosis | Amerindia | 2 años | 2 años | > 50 | No |
| Ferritina, ng/ml | Crisis vasooclusivas | Síndrome torácico agudo | Nefropatía | Vasculopatía SNC | Quelación | Hidroxiurea |
| 1,315 | Sí | Sí | No | No | – | Sí |
| < 1,000 | Sí | No | Sí | No | – | Sí |
| 1,450 | Sí | No | Sí | Sí | Deferasirox | Sí |
ND: no disponible.
Los 3 pacientes afectados de ED eran niños. La mediana de edad al diagnóstico fue de 24 meses (intervalo: 19-24 meses). Uno de ellos había recibido más de 20 transfusiones previas al alo-TPH, dado que estaba en régimen hipertransfusional como profilaxis de recurrencia tras un episodio de accidente vascular cerebral. Los 3 pacientes habían recibido tratamiento con hidroxiurea. Todos los niños habían requerido múltiples ingresos por crisis vasooclusivas y habían presentado complicaciones propias de la enfermedad: síndrome torácico agudo grave, nefropatía moderada y vasculopatía cerebral severa, respectivamente.
Las características de los pacientes se detallan en la tabla 2.
Características del trasplante alogénico de progenitores hematopoyéticosTalasemia maiorLa mediana de edad en el momento del alo-TPH fue 6 años (intervalo: 1-16 años).
Se realizó alo-TPH de donante familiar en 11 casos (9 hermanos HLA idéntico, 2 progenitores con una diferencia antigénica HLA) y en 3 casos de DNE HLA-idéntico. La fuente fue la médula ósea en todos los casos, excepto uno en que se utilizó sangre periférica y otro en que se utilizaron médula ósea y sangre de cordón umbilical.
Las características de los alo-TPH se detallan en la tabla 1.
Enfermedad drepanocíticaLa mediana de edad en el momento del alo-TPH fue 12 años (intervalo: 8-15 años). Se realizó alo-TPH de médula ósea de hermano HLA-idéntico en los 3 casos.
Las características de los alo-TPH se detallan en la tabla 1.
Evolución postrasplante alogénico de progenitores hematopoyéticosTalasemia maiorInjertoSe produjo injerto medular de los 14 pacientes trasplantados. Se observó una recuperación de la cifra de neutrófilos superior a 0,5×10e9/l en una mediana de 21 días (intervalo: 15-30 días).
Dos pacientes presentaron fallo de injerto secundario, a los 3 meses, y a los 3 años y siete meses post alo-TPH, con pérdida de la quimera del donante. Tras el diagnóstico de fallo de injerto, en ambos casos se objetivó una caída progresiva de la cifra de hemoglobina, precisando nuevamente transfusiones de hematíes. En uno de los pacientes se realizó infusión de linfocitos de donante, sin que se pudiera objetivar respuesta (tabla 3).
Características de los pacientes con quimerismo mixto
| Caso 1 | Caso 2 | Caso 3 | Caso 4 | |
| Enfermedad de base | Talasemia | Talasemia | Talasemia | Talasemia |
| Edad al alo-TPH | 8 años 10 meses | 4 años 1 mes | 13 años 7 meses | 2 años 10 meses |
| Grupo de riesgo | 3 | 3 | 2 | 1 |
| Tipo alo-TPH, fuente | Hermana, mo | Hermano, mo | Hermano, mo | Padre, sp |
| Acondicionamiento | Bu + Cy + GAT | Bu + Cy + GAT | Bu + Cy | Bu + Cy |
| Rechazo injerto | Sí | Sí | No | No |
| Evolución quimerismos | 50% D, 50% R | 100% D | 83% D, 17% R | 100% D |
| 75% D, 25% R | 100% R | 84% D, 16% R | 80% D, 20% R | |
| 60% D, 40% R | 78% D, 22% R | 72% D, 28% R | ||
| 64% D, 36% R | 78% D, 22% R | 77% D, 23% R | ||
| 62% D, 38% R | 78% D, 22% R | 60% D, 40% R | ||
| 66% D, 34% R | 79% D, 21% R | 63% D, 37% R | ||
| 46% D, 54% R | 61% D, 39% R | |||
| 13% D, 87% R | 60% D, 40% R | |||
| 23% D, 77% R | 62% D, 38% R | |||
| 25% D, 75% R | 80% D, 20% R | |||
| 23% D, 77% R | ||||
| Niveles Hb, g/dl | 6,9–7,5 | 6,2-7,2 | 15 | 11,5 |
| Electroforesis Hb | HbA2 4,5% | HbA2 3% | Normal | Normal |
| HbF 11,2% | HbF 10,8% | |||
| Plan | Transfusión hematíes cada 6 meses | Transfusión hematíes cada 2 semanas | ||
| Transfusión linfocitos de donante, no efectivo | Pendiente 2.° alo-TPH | |||
| Pendiente 2.° alo-TPH hermano HLA-idéntico |
D: donante; R: receptor.
Diez niños mantuvieron un quimerismo completo del donante y 2 presentaron un quimerismo mixto pero suficiente para mantener una cifra de hemoglobina estable sin requerimiento de transfusiones (tabla 3). Por tanto, en estos 12 pacientes la cifra de hemoglobina permaneció dentro de los intervalos de la normalidad y la electroforesis de hemoglobina se normalizó.
Enfermedad injerto contra huéspedOcho pacientes desarrollaron EICH aguda, en grado i-ii, en una mediana de 20 días post alo-TPH (intervalo: 15-30 días). Todos evolucionaron satisfactoriamente tras tratamiento con corticoides.
Tres pacientes desarrollaron EICH crónica durante el seguimiento posterior, 2 de los cuales fueron pacientes sometidos a TPH de DNE. Los órganos afectados fueron: pulmón (bronquiolitis obliterante con neumonía organizativa), piel e intestino. Fueron tratados con corticoides, añadiéndose en los pacientes con afectación pulmonar azitromicina, con mejoría progresiva y evolución favorable posterior.
Otras complicaciones precocesLas complicaciones más frecuentes en el periodo post alo-TPH inmediato fueron: mucositis relacionada con el tratamiento de acondicionamiento y neutropenia febril.
Evolución a largo plazoLa mediana de seguimiento fue de 6 años (intervalo: 1-15 años).
Los 2 pacientes con fallo de injerto entraron nuevamente en programa transfusional.
Los 12 pacientes con quimerismo estable consiguieron una cifra de hemoglobina y una electroforesis de hemoglobina normales, sin precisar nuevas transfusiones.
En el seguimiento endocrinológico se valoró el crecimiento en 10 pacientes, 5 presentaron recuperación (todos ellos catalogados como clase 1 o 2 pre alo-TPH, menores de 8 años en el momento alo-TPH, sin EICH crónica), 2 mantuvieron un crecimiento estable en percentil de peso y talla, y los 4 restantes (incluidas las 2 niñas con fallo de injerto y 2 pacientes sometidos a TPH DNE en tratamiento crónico con corticoides por EICH crónica) presentaron un empeoramiento del percentil de peso y talla respecto al que presentaban en el momento del alo-TPH.
Se detectó hipogonadismo hipogonadotropo en 5/8 pacientes de edad superior a 13 años (4 niñas y un niño).
No se detectaron otras complicaciones relevantes.
Enfermedad drepanocíticaInjertoSe produjo injerto medular de los 3 pacientes trasplantados. Se observó una recuperación de la cifra de neutrófilos superior a 0,5×10e9/l en una mediana de 21 días (intervalo: 15-30 días).
Todos presentaron un quimerismo completo del donante durante el seguimiento.
Enfermedad injerto contra huéspedDos pacientes desarrollaron EICH aguda, en grado i-ii, en una mediana de 21 tras el alo-TPH (intervalo 13-31 días). Todos evolucionaron satisfactoriamente tras tratamiento con corticoides.
No se detectó EICH crónica durante el seguimiento posterior.
Otras complicaciones precocesLas complicaciones más frecuentes en el periodo post alo-TPH inmediato fueron: mucositis relacionada con el tratamiento de acondicionamiento y neutropenia febril. Se detectó hipertensión arterial en 2 pacientes, bien controlada con tratamiento hipertensivo. No se detectaron complicaciones neurológicas.
Evolución a largo plazoLa mediana de seguimiento fue de 3 años (intervalo: 1-4 años).
Los 3 pacientes presentaron cifras de hemoglobina y electroforesis de hemoglobina normales. No presentaron nuevos episodios vasooclusivos ni otras complicaciones secundarias a la enfermedad de base. Un paciente con afectación neurológica en el momento del alo-TPH, consistente en sordera neurosensorial y hemiparesia izquierda, se mantuvo estable tras el alo-TPH, sin progresión ni regresión de la misma. Otro de los pacientes con tubulopatía secundaria ha presentado mejoría progresiva de la misma tras el alo-TPH.
En el seguimiento endocrinológico, se valoró el crecimiento en 2 pacientes, se evidenció un desarrollo pondoestatural estable, manteniendo el mismo percentil de peso y talla tras el alo-TPH.
Se detectó hipogonadismo hipogonadotropo en uno de los 3 pacientes.
DiscusiónLa TM y la ED son afecciones graves, con una esperanza de vida disminuida, que condicionan un seguimiento médico periódico y estricto, y la necesidad de tratamiento médico crónico.
Los niños afectados de TM dependen de transfusiones de hematíes cada 3-4 semanas. Como consecuencia, presentan sobrecarga férrica con riesgo de miocardiopatía secundaria o arritmias, con una esperanza de vida de apenas 10 años antes de la aparición de los quelantes de hierro2. A partir del año 1962, con la aparición de la deferoxamina subcutánea, la morbimortalidad disminuyó, permitiendo una supervivencia de 35 años en el 50-60% de los pacientes que tenían un buen cumplimiento1. Sin embargo, la dificultad en su administración, diaria, subcutánea y en infusión de 8-12 h, dificultaba el cumplimiento. Con la aparición de los quelantes orales, la deferiprona en 1980 y el deferasirox en el 2006, el cumplimiento mejoró1,2,8. En nuestra casuística, 7/14 pacientes recibieron tratamiento con deferoxamina con irregular cumplimiento, 2 con deferoxamina más deferiprona y 3 con deferasirox, con buen cumplimiento. Dado el poco tiempo de seguimiento de los pacientes con tratamiento quelante oral, no se dispone de datos fiables del impacto que la mejoría en la adhesión al tratamiento supondrá en la calidad y la esperanza de vida, y si la seguridad y la eficacia de los tratamientos orales son equiparables a los resultados obtenidos por la deferoxamina1,2,9.
El alo-TPH permite al paciente prescindir de la necesidad de transfusiones periódicas, evitando así las complicaciones secundarias de estas. En nuestra experiencia, tras una mediana de seguimiento de 6 años, 12/14 pacientes permanecen asintomáticos. Si se dispone de un hermano HLA idéntico el alo-TPH, es el tratamiento de elección. Sin embargo, el alo-TPH no está exento de riesgos: 3/14 pacientes presentaron EICH crónica y en 5 se ha detectado hipogonadismo hipogonadotropo. La aparición de los quelantes orales de hierro y un mejor seguimiento de estos pacientes han permitido una calidad y esperanza de vida superiores, que hay que sopesar frente a los riesgos de un alo-TPH8.
En 1991 se publicaron en New England Journal of Medicine los criterios de Pesaro 3 factores independientes que reducen significativamente la supervivencia y la supervivencia libre de TM tras el alo-TPH: hepatomegalia, fibrosis portal y mal cumplimiento del tratamiento quelante. Se catalogan como clase 1 los pacientes sin factores de riesgo y clase 3 aquellos con los 3 factores5,10. Diferentes estudios han demostrado un riesgo aumentado de fallo de injerto en los pacientes clase 310. Los niños que han recibido un mayor número de transfusiones, con el consecuente riesgo de alosensibilización, tienen un riesgo aumentado de rechazo y recurrencia de la enfermedad de base10. En nuestra casuística, 2 niñas de 8 y 4 años, respectivamente, catalogadas como clase 3, presentaron fallo de injerto secundario.
Históricamente, la indicación de alo-TPH estaba limitada a paciente con donante hermano HLA idéntico5. Tras los buenos resultados iniciales obtenidos en el alo-TPH de DNE HLA idéntico11, se ampliaron las indicaciones. En nuestra serie, se realizó alo-TPH de hermano HLA idéntico en 9 casos, de familiar HLA compatible en 2 casos y en 3 de DNE HLA idéntico. Actualmente, el alo-TPH continúa siendo el tratamiento de elección si se dispone de donante hermano HLA idéntico. En el caso de no disponer de este, cada centro debe sopesar los riesgos y los beneficios de tratamiento médico con transfusiones y quelantes de hierro versus el alo-TPH de DNE9. Es decir, la disyuntiva entre ofrecer un tratamiento médico crónico no curativo que se traduce en visitas frecuentes cada 3-4 semanas al hospital y la implicación del paciente en su cumplimiento, pero con expectativas de vida cada vez más prolongadas, versus un tratamiento curativo que puede ser potencialmente mortal o puede dejar al paciente con secuelas graves de por vida. Variables como la edad, el estado clínico, las transfusiones recibidas, la adhesión al tratamiento y la experiencia del centro deben ser valoradas.
La ED, a pesar de tratamientos como la hidroxiurea y las medidas profilácticas, es una enfermedad crónica que cursa con un daño orgánico progresivo. Los 3 pacientes trasplantados en nuestro centro, menores de 16 años, habían presentado ya múltiples complicaciones. Globalmente, la esperanza de vida es de 35 años1. Dada la variabilidad en la evolución clínica de esta enfermedad y la morbimortalidad asociada al alo-TPH, en 1996 se publicaron en el New England Journal of Medicine los criterios de elegilibilidad para trasplante, donde se consideraban candidatos a alo-TPH los pacientes con crisis vasooclusivas recurrentes (más de 2 episodios al año por varios años), priapismo recurrente, síntomas o signos radiológicos de afectación del SNC, nefropatía moderada-severa, síndrome torácico recurrente o afectación pulmonar moderada-severa, retinopatía y osteonecrosis6. Diferentes estudios han valorado el impacto del alo-TPH de hermano HLA idéntico en la supervivencia global (97%) y la supervivencia libre de enfermedad (85%), y el impacto en el daño cerebral y pulmonar, mostrando que en la mayoría de los casos se produce una estabilidad e incluso mejoría de ambos1,7,12,13. Por este motivo, las indicaciones de trasplante se han ido ampliando incluso a pacientes asintomáticos en el caso de disponer de hermano HLA-idéntico. En caso de no disponer de hermano HLA idéntico, los resultados actuales muestran un riesgo de mortalidad y EICH crónico en el alo-TPH de DNE del 30 y 27%, todavía muy alto1, por lo que en el momento actual la ausencia de hermano HLA idéntico es la principal barrera para la realización alo-TPH en niños con ED14.
En conclusión, el alo-TPH de hermano HLA-idéntico puede suponer la curación del niño en ambas afecciones, así como el alo-TPH de DNE en casos seleccionados de TM, sin requerimiento de nuevas transfusiones o tratamientos crónicos y sin nuevos síntomas, incluso en pacientes con quimerismo mixto estable15,16. En nuestra casuística, 2 pacientes afectadas de TM con quimerismo mixto permanecen libres de enfermedad. El mejor conocimiento de la historia natural de las hemoglobinopatías, los avances en la tipificación del HLA, el desarrollo de regímenes de acondicionamiento adecuados al riesgo del paciente y cada vez menos tóxicos, y un mejor tratamiento de soporte han permitido que los resultados del alo-TPH en el paciente afectado de hemoglobinopatía sean cada vez más satisfactorios1. La fuente de progenitores hematopoyéticos de elección es médula ósea de hermano HLA-idéntico, pero actualmente en pacientes con TM se obtienen también excelentes resultados utilizando sangre de cordón umbilical de hermano HLA-idéntico y DNE1,11,17,18. Se están desarrollando regímenes de acondicionamiento de intensidad reducida para minimizar la toxicidad, aunque los resultados, por el momento, no son suficientemente satisfactorios1,19.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
