




La miocardiopatía hipertrófica (MCH) es un trastorno genético del músculo cardíaco debido en un 60% de los casos a mutaciones de las proteínas sarcoméricas1,2, que dan como resultado la desorganización de los miocitos con fibrosis e hipertrofia miocárdica primaria.
La edad de presentación suele retrasarse hasta los 12 años. Cuando la aparece en menores de 4 años es importante descartar otras causas asociadas: glucogenosis, enfermedades mitocondriales, enfermedad de Fabry o el síndrome de Noonan2–4.
Clínicamente, la mayoría de los pacientes con MCH están asintomáticos o presentan síntomas menores, por lo que el diagnóstico se realiza fundamentalmente gracias al cribado familiar y al realizarse pruebas previa realización de actividad deportiva2,3. Entre los síntomas típicos se incluyen: disnea, dolor torácico, síncope y palpitaciones2.
La muerte súbita (MS) es el riesgo más importante de los pacientes con MCH3. En ocasiones, es la forma de presentación inicial de la enfermedad, lo que es particularmente común en adolescentes y adultos jóvenes afectados que realizan deportes de competición2,3,5,6. El ejercicio enérgico y los deportes de competición deben evitarse en los pacientes con MCH debido al aumento del riesgo de MS1,6–8.
Paciente mujer de 9 años y 50,2kg de peso (percentil>97), en clase funcional I, que consulta en dos ocasiones en el servicio de urgencias pediátricas de nuestro centro por cuadros sincopales mientras realizaba ejercicio físico (carrera). En ambos episodios refiere sensación de mareo y de falta de aire sin palpitaciones ni opresión precordial.
Entre sus antecedentes familiares su madre había sido estudiada en cardiología por episodios de dolor torácico atípico sin evidencia de cardiopatía isquémica.
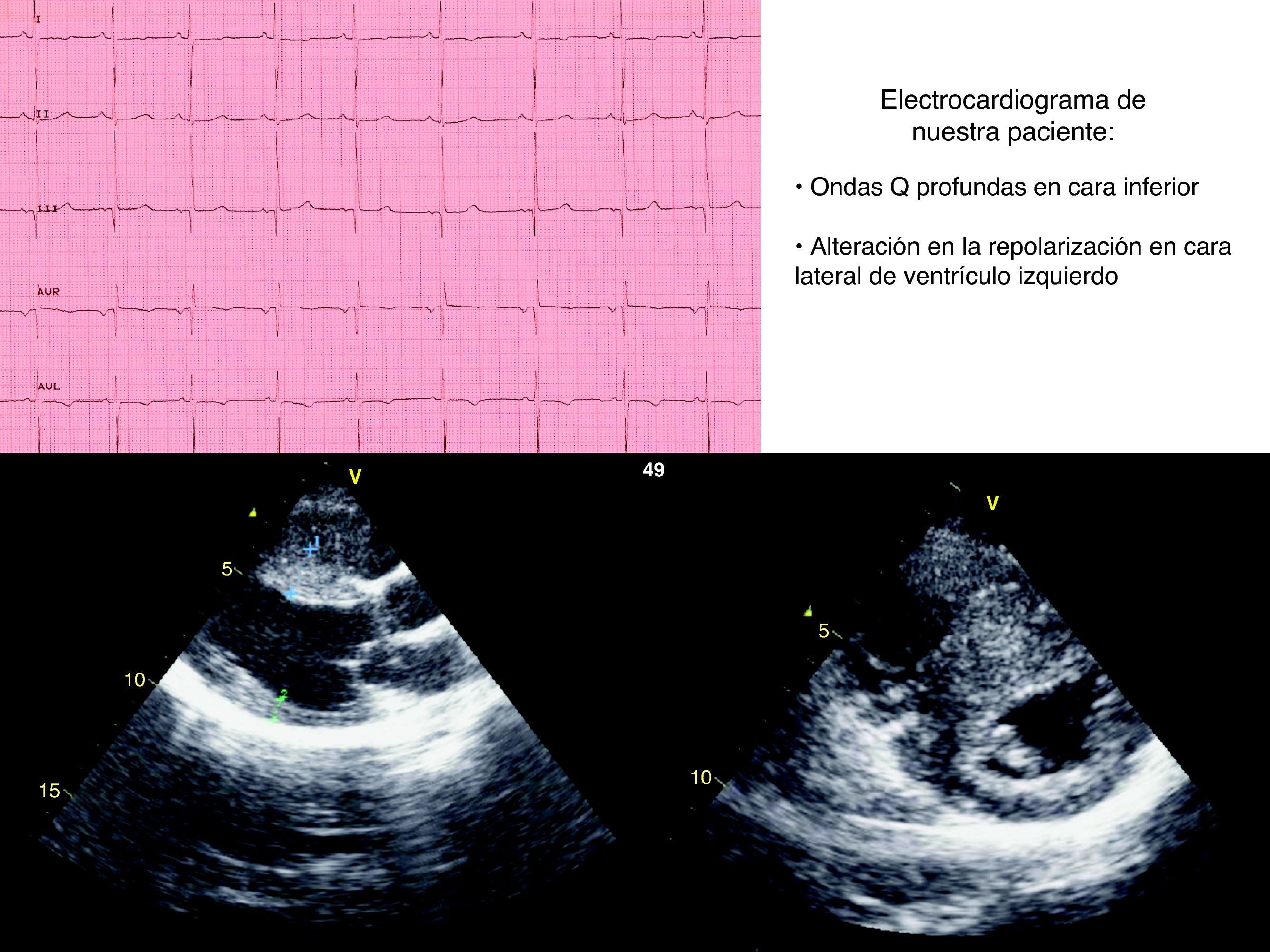
A su llegada a urgencias en ambas ocasiones se encuentra hemodinámicamente estable. En la exploración física del segundo episodio se escucha un soplo sistólico eyectivo II/VI en borde esternal izquierdo. El ECG presenta anomalías con ondas Q profundas en DIII y alteración de la repolarización en cara lateral de ventrículo izquierdo. Se realiza estudio ecocardiográfico que muestra como hallazgos hipertrofia septal (septo: 17mm) sin obstrucción al tracto de salida ventricular izquierdo (OTSVI) ni movimiento anterior de la mitral (fig. 1).
Con el diagnóstico de MCH ingresa realizándose estudios analíticos con enzimas miocárdicas, Holter-ECG y estudio de la enfermedad de Fabry, que fueron normales. Se realiza ergometría en tapiz rodante según protocolo de Bruce, con buena respuesta cronotropa y presora sin arritmias. Se solicita resonancia magnética cardíaca (cardio-RM) (fig. 2). Finalmente, se deriva a la paciente a una unidad especializada en MCH.
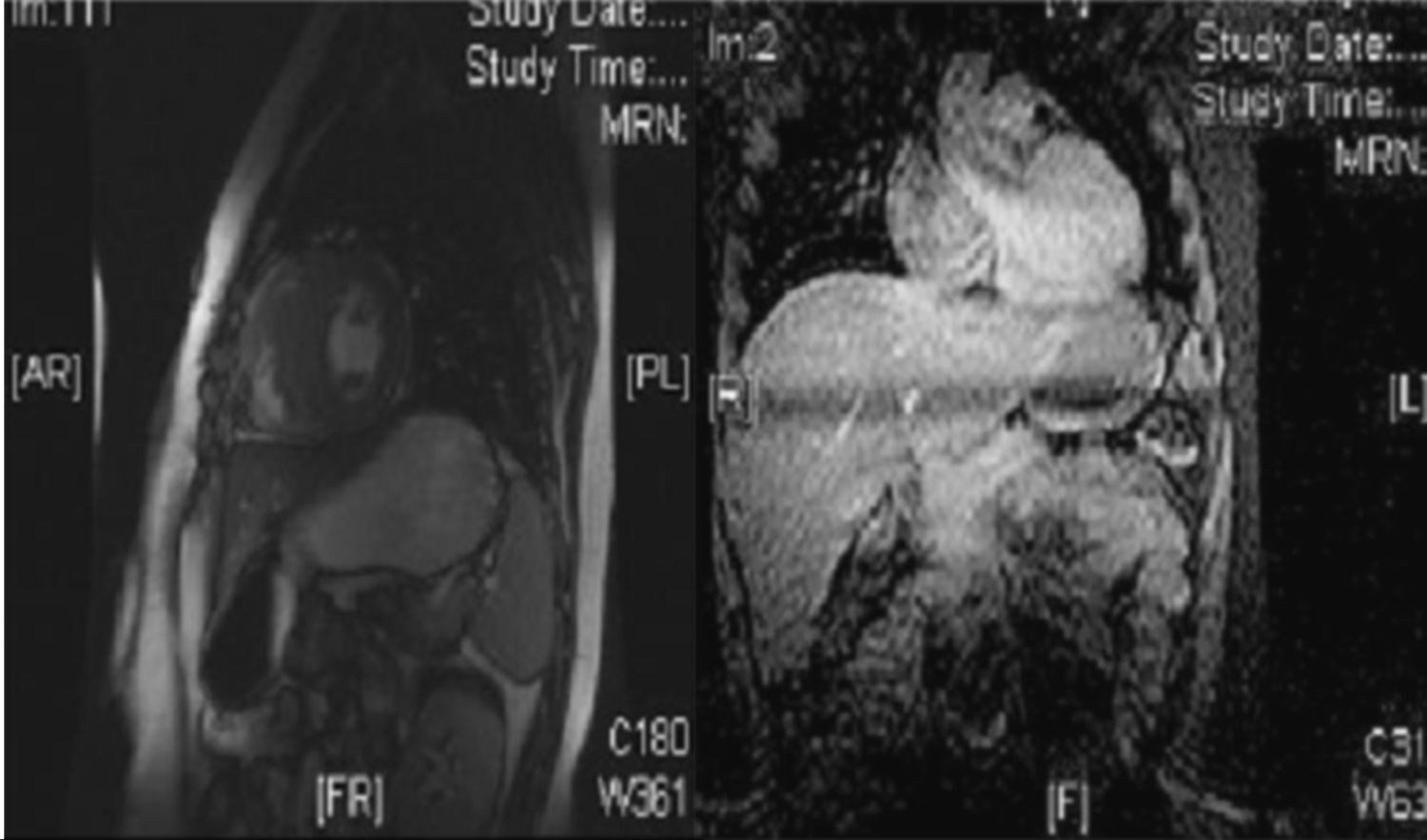
Cardio-RM. Izquierda (eje corto de ventrículos a nivel de músculos papilares): engrosamiento de septo interventricular (22mm) y de la pared posterior de ventrículo izquierdo (23mm). Derecha (corte apical cuatro cámaras): aurículas y ventrículos de tamaño y contractilidad normal con septo interventricular engrosado: 22mm.
Tras el diagnóstico se estudia a los familiares de primer grado de nuestra paciente confirmándose que la madre asimismo presentaba la enfermedad, aunque en menor grado de afectación.
Nuestro caso constituye una forma severa de inicio temprano de la enfermedad. La presencia de síncopes de esfuerzo repetidos, así como las alteraciones en el ECG, nos llevaron a la realización de estudio ecocardiográfico.
El ECG es patológico en un 75-95% de los pacientes con miocardiopatía hipertrófica3,6 mostrando múltiples alteraciones: ondas R altas en precordiales izquierdas con ondas S profundas en derechas, ondas Q profundas en derivaciones de cara inferior y ondas T negativas en cara lateral1,3,7.
La ecocardiografía continúa siendo la modalidad diagnóstica inicial para identificar a los pacientes con esta afección3. Además de medir el grosor del septo es importante evaluar la función diastólica (Doppler tisular), la dimensión de las cámaras cardíacas y la presencia de OTSVI1,2.
Los pacientes con esta enfermedad deben someterse a un registro Holter-ECG y a una prueba de esfuerzo1 con el objetivo de investigar arritmias, hipotensión inducida por el ejercicio o datos de isquemia.
El empleo de cardio-RM se está volviendo cada vez más común en la evaluación del paciente pediátrico con MCH. La existencia de realce tardío pone de manifiesto la existencia de daño miocárdico1.
El objetivo de nuestro caso es incidir en la importancia de una buena evaluación cardiológica en los síncopes de esfuerzo. Cuando la MCH se presenta de este modo constituye un factor de riesgo de MS, especialmente en jóvenes, y hace plantearse la necesidad de realización de profilaxis primaria (implantación de cardiodesfibrilador)9.
La penetrancia dentro de una misma familia y la heterogeneidad de esta enfermedad, tanto en el modo de presentación (en nuestro caso la madre de la paciente aún no había sido diagnosticada) como en el pronóstico, hacen que su manejo requiera a menudo una valoración por parte de centros especializados. Las relaciones entre el genotipo-fenotipo son complejas. Estudios de familias con MCH han demostrado la presencia de variabilidad clínica entre individuos con la misma mutación detectándose síntomas leves en un familiar y un inicio precoz de la enfermedad en otro, lo que complica el consejo genético en esta enfermedad10.
