



El síndrome de Allgrove (triple A) es una enfermedad autosómica recesiva rara. La tríada clásica incluye insuficiencia adrenal congénita debida a resistencia a la ACTH, acalasia del cardias y alacrimia. Se asocia a anomalías neurológicas, como neuropatía autonómica, sensitiva y motora, sordera, retraso mental, parkinsonismo y demencia. El gen responsable es el AAAS o ADRACALIN, que codifica una proteína llamada ALADIN. Se presenta un caso de un varón de 19 años, valorado con 10 años en nuestro servicio ante la sospecha de enfermedad de depósito. Presenta retraso mental leve y del lenguaje, voz hipernasal, neuropatía sensitivo-motora con afectación autonómica y semiología de paraparesia espástica. Alacrimia. Reflujo gastroesofágico y acalasia. El estudio molecular demostró 2 mutaciones, la p. Tyr 19 Cys no descrita, y la IVS14+1G-A.
Allgrove syndrome (triple A) is a rare autosomal recessive disease. The classic triad includes, congenital adrenal insufficiency due to ACTH resistance, achalasia of the cardia and alacrimia. Neurological abnormalities are associated with autonomic neuropathy, sensory and motor defects, deafness, mental retardation, Parkinsonism and dementia. The gene responsible is the ADRACALIN or AAAS encoding a protein called ALADIN. We report a case of a 19 year-old male, assessed when he was 10 years old in our department due to suspected storage disease. Mild mental and language retardation, hypernasal voice, sensory-motor neuropathy with autonomic involvement and signs of spastic paraparesis, alacrimia. gastroesophageal reflux, and achalasia. Molecular studies showed to mutations, the undescribed p.Tyr 19 Cys, and IVS14 +1 G.
El síndrome de Allgrove (triple A) es una enfermedad autosómica recesiva rara, descrita en el año 1978, por Allgrove et al1. La tríada clásica incluye insuficiencia adrenal congénita (debida a resistencia a la ACTH), acalasia del cardias y alacrimia (deficiente producción de lágrimas). Se manifiesta en las 2 primeras décadas mediante deficiencia glucocorticoidea, incluyendo crisis hipoglucémicas y shock. Se asocia a alteraciones neurológicas2, como neuropatía autonómica, sensitiva y motora, sordera, déficit cognitivo, parkinsonismo y demencia, por lo que algunos autores lo designan como síndrome de las 43.
Se presenta un caso de un varón de 19 años, valorado con 10 años en nuestro servicio ante la sospecha de enfermedad de depósito. Presenta retraso mental leve y del lenguaje, voz hipernasal, neuropatía sensitivo-motora con afectación autonómica y semiología de paraparesia espástica. Alacrimia. Reflujo gastroesofágico y acalasia. El estudio molecular demostró 2 mutaciones, la p. Tyr 19 Cys no descrita, y la IVS14+1G-A4.
En 1996, se demostró que la enfermedad está ligada a la banda cromosómica 12q13. En el 2000, se identificó el gen responsable: AAAS o ADRACALIN, que codifica una proteína de 547 aminoácidos, llamada ALADIN (de alacrimia, acalasia, insuficiencia adrenal y alteración neurológica), implicada en regular en transporte nucleocitoplasmático, cuya expresión en estructuras neuroendocrinas, gástricas y cerebrales desempeña un papel importante en el mantenimiento y desarrollo de determinados tejidos. Se postula que parte de la patología del síndrome se debe a una pérdida colinérgica progresiva en todo el organismo.
La enfermedad es genéticamente muy heterogénea5 y existen algunos casos descritos de pacientes diagnosticados de síndrome de Allgrove sin mutaciones en el gen AAAS, por lo que pudiera haber otros genes implicados. Por ello, en ocasiones, los pacientes son diagnosticados de otros procesos.
Caso clínicoVarón de 19 años, valorado en genética clínica a los 10 años, derivado de Neurología Infantil, por sospecha de enfermedad de depósito.
Antecedentes familiares: madre y abuela materna tuvieron 2 abortos. Padre y un hermano del padre presentan miopía severa. Antecedentes personales: producto de tercer embarazo de padres de 28 años. No consanguinidad. Parto a término, eutócico normal. No reanimación al nacer. Peso al nacer: 3.550g. Periodo perinatal normal. Adenoidectomizado. Diagnosticado a los 4 años de astigmatismo y ojo vago, con fondo de ojo normal. Presentó alacrimia a los 9 años. Se apreciaron problemas de regurgitaciones frecuentes y se pudo demostrar que estaba afectado de reflujo gastroesofágico y acalasia a la misma edad. Tiene varias dilataciones realizadas. Se detectó retraso en el desarrollo en los primeros años de vida, manifestado fundamentalmente por retraso en el lenguaje. Valorado por Neurología Infantil a los 9 años y diagnosticado de síndrome de paraparesia espástica asociada a signos de neuropatía sensitivo-motora, desmielinizante y axonal, de predominio distal, y a signos de afectación autonómica.
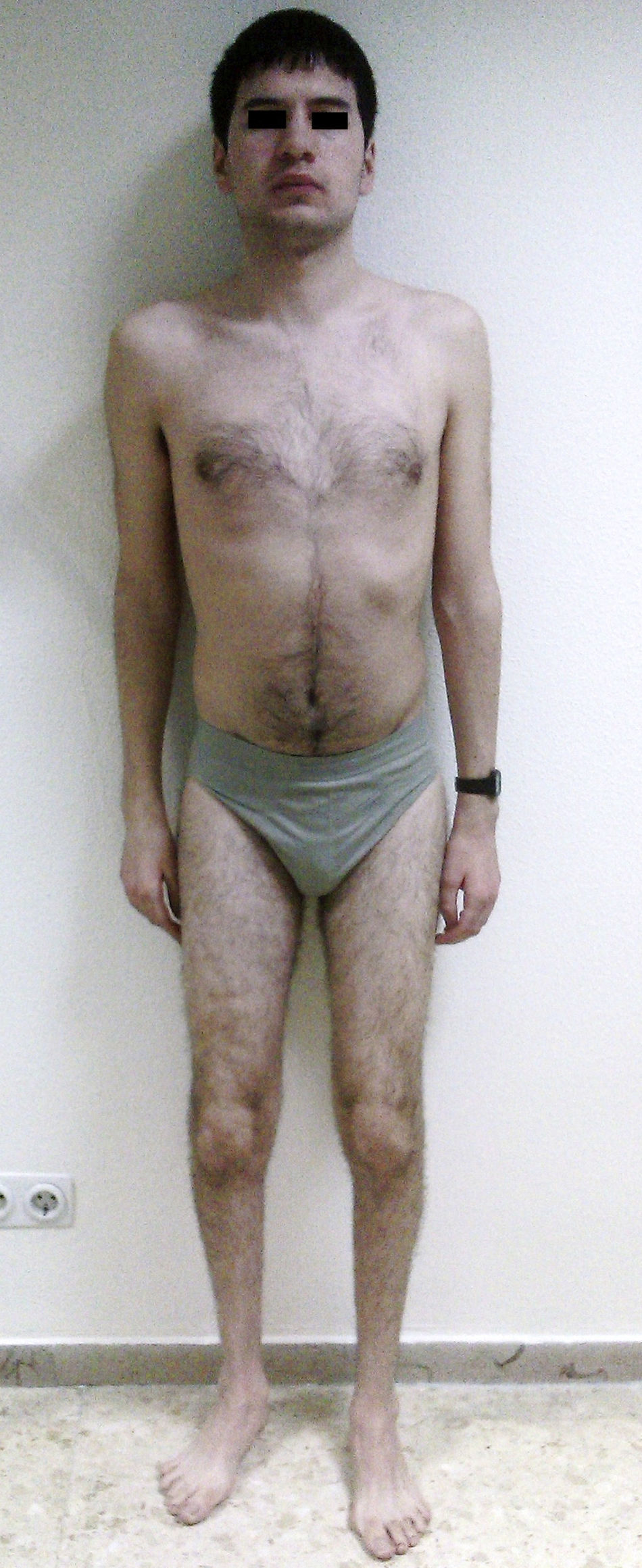
A los 19 años presenta déficit intelectual y a la exploración: peso 80kg, talla 1,77m, PC 58,8cm. Normocefalia, hendiduras palpebrales normales, raíz nasal ancha, filtro corto con labio superior evertido y lengua geográfica. Resto de facies, normal. La exploración sistemática por aparatos es normal. Presenta hipotonía axial, escasa fuerza muscular e hipertonía, de predominio distal, con escaso relieve de masas musculares, así como espasticidad e hiperreflexia y problemas de coordinación-secuenciación motriz en miembros superiores (fig. 1). Retracción aquílea bilateral, así como pies cavos bilaterales, con disposición en cavo y equino varo del izquierdo. Se aprecia fosita sacra. Cicatriz queloide supraumbilical de 6,5cm.
En los exámenes complementarios se aprecian sialotransferrinas séricas, ácidos orgánicos en orina, normales. El estudio en la biopsia muscular y de esófago de la mutación A → G en el nucleótido 3243 asociada a MELAS y el estudio de deleciones a nivel mitocondrial (por técnica Southern y amplificación del ADN humano de la región rARN 16S-NDI) fue negativo. Presenta lentificación difusa en el EEG. En el estudio neurofisiológico muestra potenciales de amplitud reducida en el nervio peroneo derecho con velocidad de conducción motora en los límites bajos de la normalidad y discretamente reducida en los miembros superiores. El cariotipo en sangre periférica por bandas GTG (550 bandas) fue normal, con una fórmula cromosómica 46, XY, inv. (9) (p11q13). Se hizo FISH para la región 22q11.2 (Tuple1×2), normal. Ante la sospecha diagnóstica, se realizó un estudio molecular de síndrome de Allgrove, realizado mediante amplificación de PCR y secuenciación de los 16 exones y de las regiones intrónicas flanqueantes del gen AAAS (ADRACALIN), que demostró 2 mutaciones, una en cada alelo: la p.Tyr19Cys (no descrita previamente) y la IVS14+1G-A. El estudio molecular del gen AAA en los progenitores demostró que la madre es portadora heterocigota de la mutación IVS 14+1G>A y el padre, de la mutación C.56A>G (p.Tyr19Cys).
DiscusiónLos hallazgos clínicos en familias con mutaciones en el gen AAAS son diversos y abarcan varios sistemas, lo que unido a la gran variedad clínica de presentación dan lugar a un diagnóstico tardío, como en el caso que presentamos.
Se han descrito cerca de 200 casos en todo el mundo, mostrando gran variabilidad tanto en la severidad como en las manifestaciones clínicas. La incidencia actual es difícil de determinar, debido a la gran variabilidad de presentación, pues algunos pacientes mueren sin diagnóstico en la infancia (debidas a crisis adrenales) y hay casos leves que no son diagnosticados hasta la edad adulta. Los síntomas iniciales suelen manifestarse en la infancia temprana. Se suele presentar como crisis hipoglucémica secundaria a déficit de glucocorticoides. La edad de comienzo de los síntomas es muy variable. La primera manifestación de la enfermedad en la infancia es la alacrimia, acompañada en el 75% de los pacientes de acalasia, que puede aparecer desde los 6 meses hasta el principio de la edad adulta, sin presentar ningún otro rasgo del síndrome de Allgrove. En nuestro caso, se manifestó en la infancia temprana. La insuficiencia adrenal suele comenzar después que la disfagia y se desarrolla gradualmente a lo largo de las 2 primeras décadas de la vida. Puede variar su expresión, pudiendo cursar únicamente como astenia, diagnosticándose erróneamente como fatiga muscular. El caso que reportamos, actualmente presenta una adecuada función suprarrenal, con adecuados niveles de ACTH, no mostrando indicios de insuficiencia glucocorticoidea. No obstante, la progresión desde una función suprarrenal normal hacia la insuficiencia ha sido documentada en numerosos individuos.
Los trastornos neurológicos tienen un curso progresivo y pueden afectar el sistema nervioso central, periférico o autónomo, mostrando manifestaciones muy heterogéneas y variadas2. El síntoma más frecuente es el trastorno disautonómico, que incluye hipotensión ortostática, disfunción vesical, diarrea o estreñimiento, disfunción sexual y dishidrosis. Nuestro paciente presenta un retraso del desarrollo, que es un hallazgo común en la población pediátrica, y aún no se sabe si se debe a un rasgo propio del síndrome o si es debido a una secuela de algún episodio de hipoglucemia. Los hallazgos neurológicos más comúnmente descritos abarcan hiperreflexia, disartria, voz hipernasal con insuficiencia velopalatina y ataxia, presentes en nuestro paciente. Otros hallazgos, como la retracción aquílea y los pies cavos, han sido descritos en otros pacientes afectados de síndrome de Allgrove.
El examen de la piel de estos pacientes suele revelar hiperpigmentación, hiperqueratosis y grietas palmo-plantares.
Otros hallazgos que se pueden encontrar en estos pacientes son: lengua fisurada, pectus excavatum, amiotrofia distal y amiotrofia de miembros superiores, con afectación predominante de la zona cubital de la mano.
En cuanto a los hallazgos electrofisiológicos, cabe destacar una disminución de la amplitud del potencial de acción en de los miembros inferiores en relación con alteraciones axonales.
Este síndrome es poco conocido y muchas veces los pacientes permanecen largo tiempo sin diagnóstico correcto.
Señalamos la importancia del conocimiento de síndrome por el pediatra, pues para realizar un diagnóstico precoz, debe hacerse análisis molecular del gen AAAS en pacientes que presenten uno o 2 de los 3 síntomas principales3,6.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
