




La microdeleción 22q11 es una deleción genética frecuente con variabilidad fenotípica amplia. Engloba una serie de síndromes, entre los que destaca el síndrome de DiGeorge. Las manifestaciones clínicas más frecuentemente descritas son malformaciones cardiacas, una facies característica, malformaciones palatinas, hipoparatiroidismo, inmunodeficiencia secundaria a hipoplasia tímica, retraso psicomotor y alteraciones psiquiátricas.
Dentro de los signos producidos por la enfermedad, es frecuente la insuficiencia respiratoria de causa multifactorial.
Las malformaciones de la vía aérea son frecuentes, aunque en la mayoría de los pacientes son leves, por lo que no suelen producir sintomatología. Sin embargo, en algunos casos pueden condicionar clínica respiratoria grave.
Los casos clínicos presentados ilustran la importancia de la valoración precoz de la vía aérea mediante fibrobroncoscopia en pacientes con microdeleción 22q11 que presentan dificultad respiratoria recurrente.
The 22q11 deletion syndrome is a frequent contiguous-gene deletion syndrome. This disorder has a broad spectrum of phenotypic manifestations. It includes various syndromes such as DiGeorge syndrome.
The most frequent clinical manifestations are congenital cardiac defects, characteristic facies, palate malformations, hypoparathyroidism, immunodeficiency due to thymic hypoplasia, growth retardation, and behavioural and psychiatric problems.
Among the symptoms observed, many patients suffer from respiratory insufficiency or failure. The origin is often multifactorial. Structural airway abnormalities are frequently found in this syndrome. In many of these patients the malformation is mild or non-existent, and remains asymptomatic. However, in some cases it can cause a severe respiratory insufficiency, being diagnosed when other disorders are ruled out.
These cases illustrate the importance of early visualisation of the airway by fibrobronchoscopy in the management of the patient with 22q11 deletion syndrome who has recurrent respiratory difficulties.
La microdeleción 22q11 está implicada en una serie heterogénea de fenotipos, como los síndromes de DiGeorge, CATCH 22 y Cayler1,2. Constituye la alteración cromosómica más frecuente (1:4.000 recién nacidos) en el ser humano, siendo el 93% de los casos mutaciones de novo3-6. Las manifestaciones clínicas más frecuentes son malformaciones cardiacas, microcefalia y facies característica: pabellones auriculares pequeños y contorno superior recto, fisuras palpebrales antimongoloides y telecantus, disfunción velopalatina, con o sin paladar hendido, y labio leporino. También puede asociarse a hipoparatiroidismo, inmunodeficiencia por hipoplasia tímica, retraso psicomotor y alteraciones psiquiátricas en la edad adulta1-3,7-9.
Presentamos los casos clínicos de 4 niñas con patología respiratoria y cardiovascular asociada a microdeleción 22q11, con el propósito de destacar el papel diagnóstico de la fibrobroncoscopia (FBC) en este tipo de pacientes.
Observación clínicaCaso 1Niña de 4 años con diagnóstico de comunicación interauricular e interventricular (CIV) al nacimiento. Tiene fenotipo dismórfico, con coloboma de iris derecho, hipertelorismo, anteversión de pabellones auriculares, platibasia, micrognatia e insuficiencia velopalatina. Asocia aplasia de timo, y peso y talla bajo percentil3. El estudio confirmó, mediante hibridación fluorescente in situ (FISH), la microdeleción 22q11.
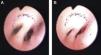
Al mes de vida presentó un cuadro compatible con broncoaspiración, consistente en dificultad respiratoria grave, con desaturación y acidosis respiratoria, y precisó ingreso en unidad de cuidados intensivos pediátricos (UCIP) e intubación, que resultó dificultosa por micrognatia y glotis desplazada anteriormente. Permaneció con ventilación mecánica durante 18 días. Pocas horas tras la extubación presentó ruido piante inspiratorio y signos de distrés, realizándose FBC urgente, que objetivó sendas membranas subglótica y traqueal amplias, con estenosis grave (obstrucción del 70 y el 90% de la luz, respectivamente) (fig. 1A y B). Se realizó split cricotiroideo urgente ampliado hasta el tercer y cuarto anillos traqueales, manteniéndose 2 semanas más intubada y conectada a ventilación mecánica. La FBC de control mostró estenosis subglótica leve, de unos 3cm de longitud, y malacia traqueobronquial. Posteriormente, se confirmó la sospecha de reflujo gastroesofágico (RGE) grave con afectación y trastorno de la deglución, indicándose funduplicatura y gastrostomía percutánea. En este periodo se aislaron Streptococcus pneumoniae y Stenotrophomona maltophilia (S. maltophilia) en el aspirado broncoalveolar, que fueron tratados según antibiograma. A los 4 meses de edad recibió el alta de la UCIP, con oxigenoterapia en gafas nasales. Actualmente, tiene 4 años de edad, presenta cuadros broncoobstructivos ocasionales leves y no precisa oxigenoterapia.
Caso 2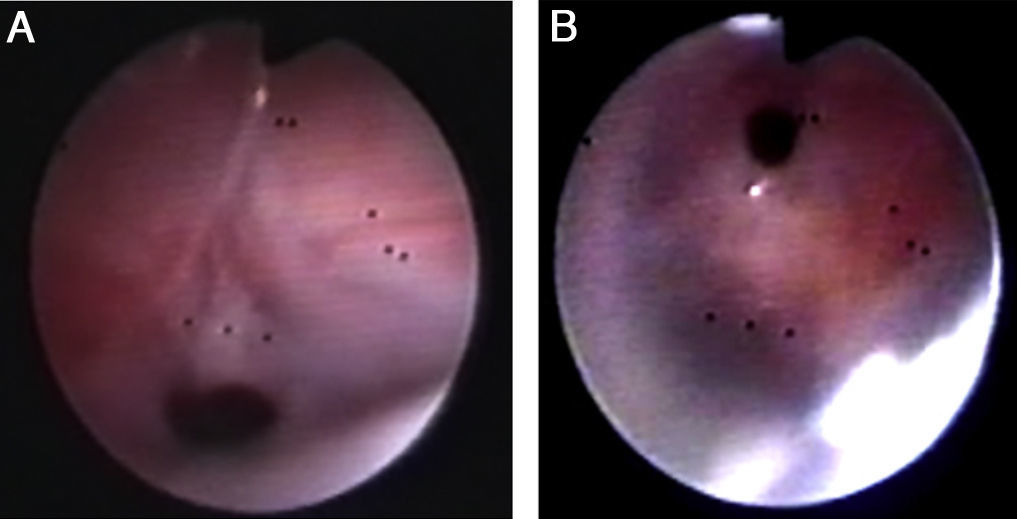
Recién nacida diagnosticada de síndrome de DiGeorge (microdeleción 22q11 en FISH), malformación de Arnold-Chiari tipo II, con mielomeningocele lumbosacro intervenido, inmunodeficiencia celular parcial con inmunidad humoral normal y retraso ponderoestatural (peso y talla<P3). Es remitida a nuestro centro para cirugía cardiaca de truncus arterioso, con foramen oval permeable y CIV, con clínica de insuficiencia cardiaca congestiva. Con 25 días de vida ingresó en UCIP tras la corrección completa de su cardiopatía. En el postoperatorio presentó hipoxemia, hipercapnia y signos de hipertensión pulmonar secundarios a shunt intracardiaco residual.
Desarrolló neumonía, con cultivos de lavados broncoalveolares positivos a Stenotrophomonas maltophila y Pseudomonas aeruginosa, y persistencia de imágenes radiológicas de bronconeumonía, bullas y atelectasia en el vértice pulmonar derecho. Se consigue extubación definitiva a los 3 meses de edad tras varios intentos fallidos, en los que se descartaron alteraciones del movimiento diafragmático y lesión troncoencefálica, precisando CPAP intermitente. Ante la persistencia de la atelectasia del lóbulo superior derecho, se realizó FBC, objetivándose una compresión pulsátil en el tercio inferior de la tráquea que ocluía parcialmente la raíz de los bronquios principales, fundamentalmente el derecho, durante los movimientos respiratorios. Una angio-TC torácica confirmó la compresión bronquial por el arco aórtico derecho, por lo que a los 5 meses de vida se indicó aortopexia del arco y la aorta descendente, comprobándose la liberación mediante broncoscopia intraoperatoria.
Tras la intervención, presentó episodios de broncoespasmo. Se realizó una nueva FBC, descartando compresión y observándose estenosis del bronquio segmentario anterior del lóbulo superior derecho, así como broncomalacia generalizada, además de granulomas por roce en ambas caras del bronquio del lóbulo inferior derecho (fig. 2A y B). Mejoró tras recibir corticoides sistémicos y se extubó al mes de la intervención. La evolución posterior fue desfavorable, precisando ventilación mecánica alterna con CPAP nasal por persistencia de broncoespasmo. Otra FBC a los 9 meses objetivó compresión pulsátil de bronquio principal derecho, con oclusión casi total de la luz bronquial, y compresión extrínseca pulsátil del bronquio izquierdo que ocluía parcialmente la luz bronquial, siendo la malacia de cartílagos mayor que en controles previos. Dos días más tarde presentó broncoespasmo grave, con desaturación y bradicardia extrema, que no respondió a maniobras de reanimación cardiopulmonar, y falleció.
Caso 3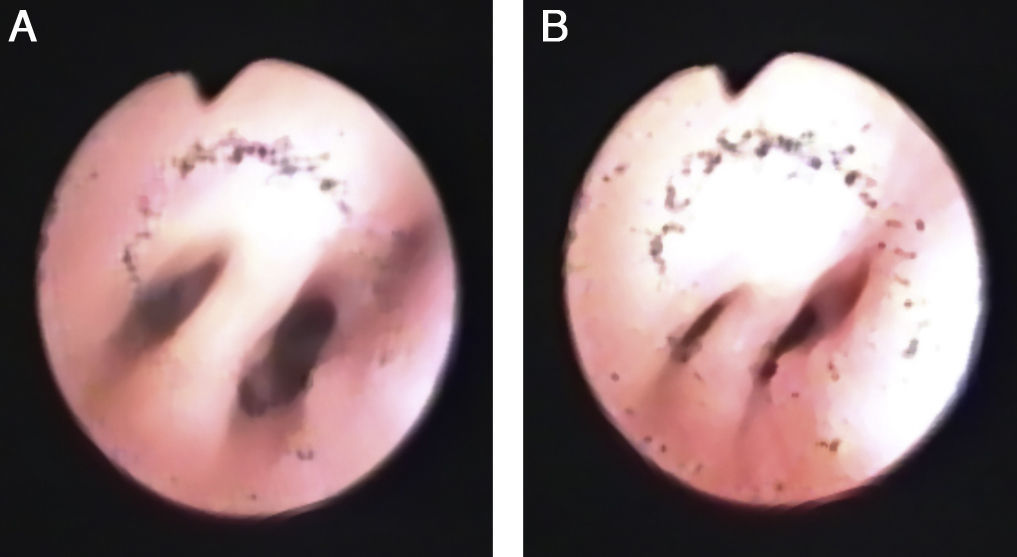
Niña de 9 meses, con antecedentes de hernia diafragmática congénita y parálisis diafragmática derechas, que precisó plicaturas (3) en el periodo neonatal. Portaba traqueostomía desde el periodo neonatal. Asociaba tetralogía de Fallot, facies peculiar, retraso ponderoestatural, RGE y colestasis neonatal. El estudio genético reveló microdeleción 22q11 (técnica de FISH).
Se realizó una intervención cardiaca correctora a los 9 meses, presentando dificultad respiratoria y crisis de broncoespasmo, tratada con corticoides y broncodilatadores inhalados, azitromicina y montelukast por vía oral. En las radiografías de tórax seriadas se apreciaba una elevación del hemidiafragma derecho, indicándose una gammagrafía de perfusión pulmonar con macroagregados de albúmina-Tc 99m, que reveló una gran asimetría de perfusión pulmonar (pulmón derecho: 36,9%, pulmón izquierdo: 63,1%) (fig. 3A y B).
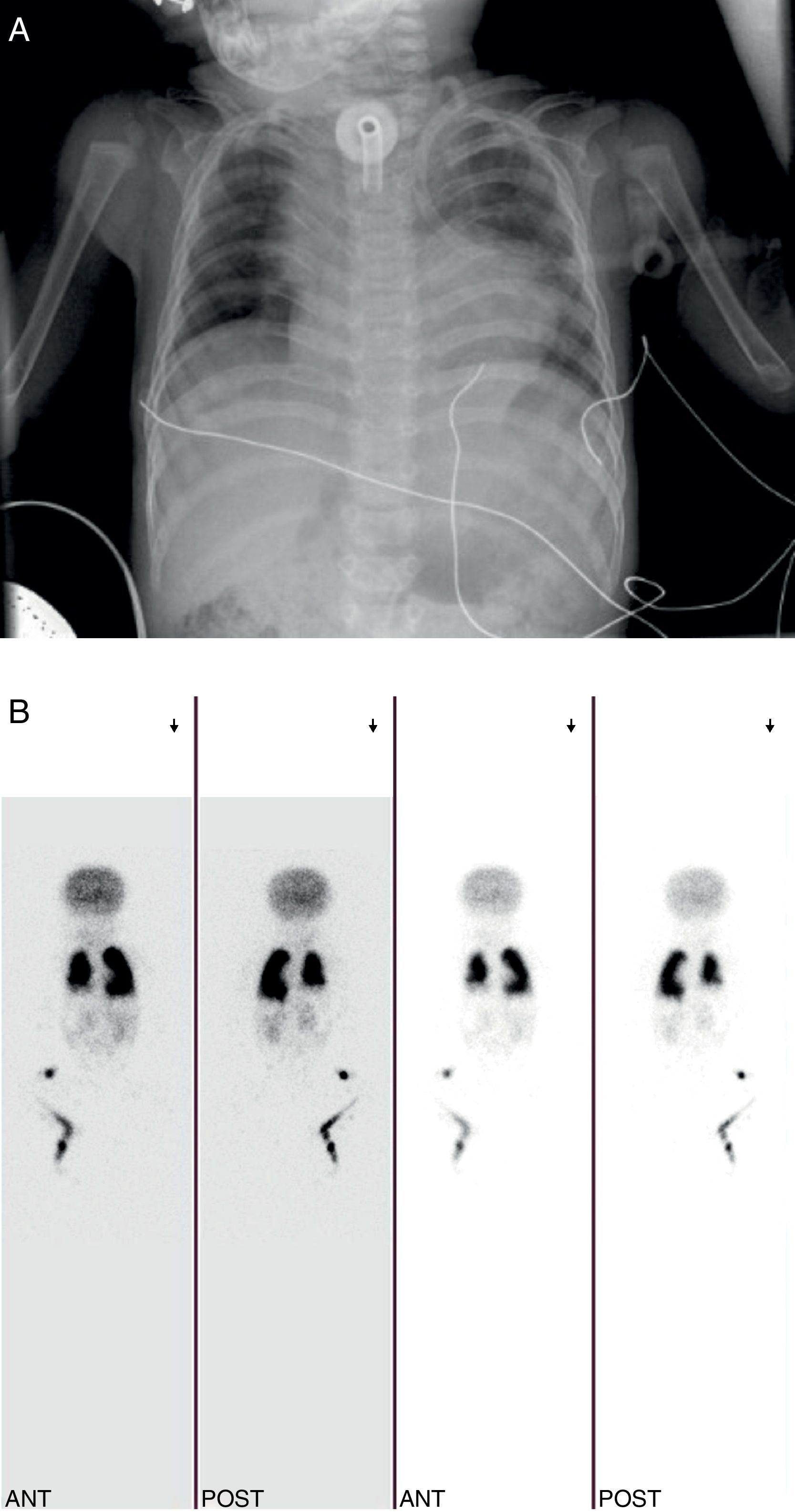
Fue dada de alta con ventilación mecánica domiciliaria, pudiendo retirarse la traqueostomía a los 2 años de edad.
Actualmente, la paciente tiene 3 años y precisa oxigenoterapia domiciliaria intermitente.
Caso 4Niña con facies peculiar (frente ancha, hipertelorismo y micrognatia), diagnosticada al nacimiento de interrupción del arco aórtico y CIV amplia, que precisaba aportes elevados de calcio.
Se confirmó la sospecha de microdeleción 22q11(FISH) y a los 15 días se intervino mediante cirugía de Norwood y conducto de Sano, observándose ausencia de timo. En el postoperatorio presentó hipoxemia marcada y patrón radiológico de isquemia pulmonar, con ausencia de flujo en el conducto de Sano, reinterviniéndose 24 h después (fístula carótido-pulmonar). Tras ello, mejoró hemodinámicamente y mantuvo la SatO2 alrededor del 75%.
Se extubó al mes de la intervención, manteniéndose con CPAP nasal. La radiografía de tórax mostraba una elevación del hemidiafragma derecho, descartándose parálisis del mismo. Se realizó una FBC, que mostró compresión extrínseca pulsátil del tercio inferior de la tráquea y el bronquio principal izquierdo. En este contexto, presentó un aumento progresivo de la dificultad respiratoria y una mayor hipoxemia, siendo reintubada. A pesar de la asistencia respiratoria, el óxido nítrico y los fármacos vasoactivos, persistían la hipoxemia severa y la inestabilidad hemodinámica. Se realizó una angio-TC torácica, que mostró una obstrucción casi completa de la arteria pulmonar derecha, posiblemente secundaria a trombosis (fig. 4). Dada la situación de isquemia pulmonar, se realizó trombólisis con rTPa y heparina por vía intravenosa, con mejoría importante de la oxigenación y estabilización hemodinámica en las horas siguientes, pudiéndose retirar la asistencia respiratoria y el soporte vasoactivo en los días posteriores. El estudio hematológico reveló una enfermedad tromboembólica por resistencia a la proteína C activada, debido a la mutación Arg 506 Gln (en heterocigosis) del factor V de Leiden. Se pautó profilaxis con ácido acetilsalicílico y heparina subcutánea ante situaciones de riesgo (cirugía, catéteres centrales).
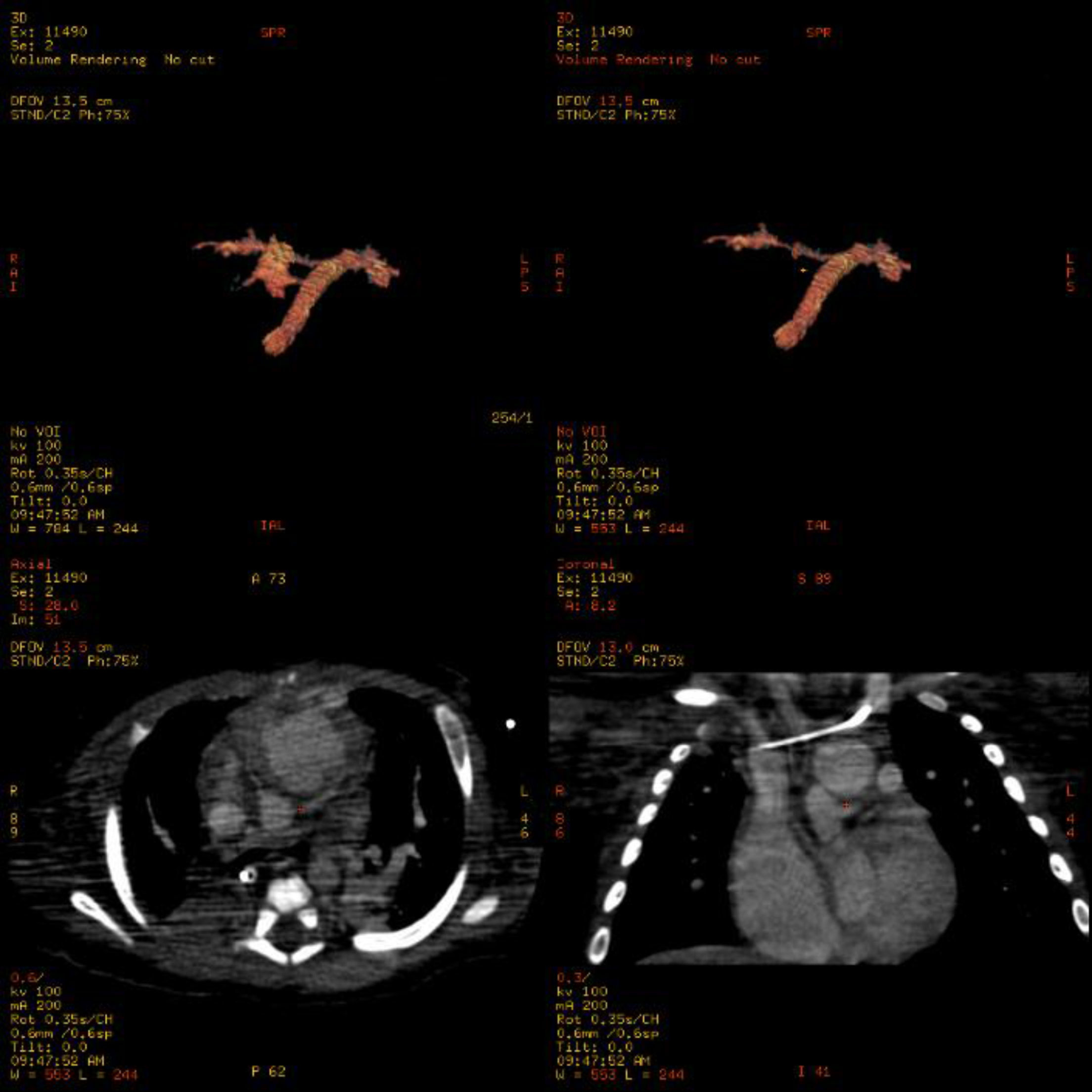
A los 4 meses de edad se realizó una cirugía de Glenn y una trombectomía en la arteria pulmonar derecha. El postoperatorio cursó sin complicaciones y la evolución posterior fue favorable. Actualmente, la paciente tiene un año de edad y no precisa oxigenoterapia.
DiscusiónEl síndrome de microdeleción 22q11 engloba un amplio espectro de alteraciones y malformaciones debidas a la alteración del desarrollo de las bolsas faríngeas1,2,5,7,9.
La FISH identifica un 95% de los casos. Además, pueden aparecer manifestaciones clínicas no asociadas a la microdelección4,7. En nuestra serie, una paciente asociaba mutación del gen de la protrombina situado en la región 1q23, mielomeningocele y malformación de Arnold-Chiari, que podrían corresponder a patología asociada coincidente o constituir manifestaciones excepcionales de la microdeleción 22q117. No existen diferencias entre etnias ni sexos5,7, aunque los 4 casos presentados son mujeres.
La manifestación clínica más frecuente es la cardiopatía (hasta 75%), en su mayoría de tipo conotruncal3,5,7,8. Las 4 pacientes presentaban cardiopatía y 3 de ellas requirieron intervención quirúrgica precoz. El segundo caso presentaba malacia bronquial grave, con compresión extrínseca pulsátil por aorta derecha promitente, que persistió a pesar de aortopexia, contribuyendo la cardiopatía a la grave insuficiencia respiratoria y al fallecimiento.
En el cuarto caso, la patología tromboembólica condicionó una inestabilidad hemodinámica grave.
Existen anomalías palatinas (incompetencia velofaríngea y paladar hendido submucoso) y retrognatia en 70% de los casos. Ello condiciona intubaciones difíciles, necesidad de traqueostomía y alteraciones fonatorias8,10. Los problemas de deglución son frecuentes, con regurgitación y aspiraciones secundarias5. El RGE también constituye una anomalía relativamente frecuente, pudiendo provocar neumopatía por reflujo3,8; el primer caso requirió intervención quirúrgica por este motivo.
La laringe deriva del cuarto y el quinto arcos braquiales, cuya proximidad embriológica a la tercera y la cuarta bolsas faríngeas podría explicar la presencia de alteraciones laríngeas8,9,11-13. La anomalía más frecuente descrita es la atresia laríngea de tipo III o membrana laríngea anterior, ya que es la más leve del espectro14. Esta produce una obstrucción parcial a nivel de la glotis. El primer caso presentaba una membrana subglótica.
Las notificaciones de malformaciones de la vía aérea inferior no son frecuentes, pero pueden condicionar clínica respiratoria grave e impedir la retirada de asistencia respiratoria. Sein et al.13 comparan la vía aérea de pacientes con síndrome de DiGeorge y controles sanos, describiendo en los 9 primeros cartílago tiroides pequeños y de morfología anormal, tráquea corta (menor número de anillos traqueales) y fístula traqueoesofágica.
También se ha descrito malacia laríngea, traqueal y bronquial, acompañadas o no de membrana laríngea con una frecuencia que se ha estimado en un 3% de los pacientes 22q1114. La malacia puede ser congénita o adquirida. Es una complicación de las cardiopatías congénitas con un shunt izquierda derecha importante. Sin embargo, en el caso de la paciente con truncus arterioso, la compresión de la aorta prominente fue la causa de la broncomalacia grave. Este mecanismo de compresión extrínseca de la vía aérea se ha descrito en pacientes con microdeleción y cardiopatías tipo seudotruncus (Fallot extremo), por la aorta o por las arterias colaterales aortopulmonares que presentan15. La malacia es más marcada en pacientes que precisan intubaciones prolongadas. La FBC es un método diagnóstico no invasivo eficaz para detectar malformaciones de la vía aérea, por lo que debe realizarse precozmente en casos de insuficiencia respiratoria de etiología dudosa. Otra condición agravante es la inmunodeficiencia de células T que se observa hasta en un 77% de los pacientes. La gravedad depende del grado de hipoplasia tímica, habitualmente leve-moderada, por lo que no son frecuentes las infecciones oportunistas, aunque sí suelen aparecer infecciones respiratorias recurrentes3-6. En nuestra serie varias pacientes presentaron neumonía nosocomial, asociadas a ingreso en la UCIP, intubación prolongada y antibioterapia de amplio espectro. Podemos concluir que en los pacientes con microdeleción 22q11, además de las manifestaciones más frecuentes (cardiopatía, alteraciones craneofaciales e inmunodeficiencia), se debe evaluar la posibilidad de patología respiratoria, cuya causa es multifactorial. Destacan la neumopatía aspirativa y las malformaciones de las vías aéreas, que pueden ser primarias o secundarias a compresión extrínseca de la vía aérea o a intubación prolongada. En este tipo de patología, la FBC tiene gran utilidad en el diagnóstico precoz.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
