



Introducción
El síndrome de Ondine consiste en un síndrome de hipoventilación central congénita secundario un trastorno del sistema nervioso central (SNC) en el cual el control autonómico de la respiración está ausente o se encuentra deteriorado en ausencia de enfermedad del tronco del encéfalo, neuromuscular, pulmonar, metabólica o cardíaca que lo justifique 1. Actualmente se cifra la incidencia de esta entidad en un caso por cada 200.000 nacimientos 2; aunque es muy probable, dado el desconocimiento de este trastorno entre la comunidad médica y la variabilidad de formas clínicas, que se trate de una enfermedad infradiagnosticada 3. El objetivo de esta revisión, fruto de una exhaustiva búsqueda bibliográfica y de la propia experiencia adquirida, es dar unas pinceladas al lector que permitan un diagnóstico precoz y un tratamiento adecuado, hechos que marcarán el pronóstico de esta entidad. Por ello se ha presentado expresamente en forma de guía práctica. Como veremos a continuación, una vez establecido el diagnóstico e iniciado un soporte ventilatorio adecuado, el paciente deberá ser atendido desde un enfoque multidisciplinar; por lo cual puede ser aconsejable la remisión a un centro de referencia.
Fisiopatología
Los pacientes afectados de síndrome de Ondine presentan una alteración difusa del sistema nervioso vegetativo que se manifiesta principalmente por un déficit de sensibilidad a la hipercapnia y una sensibilidad variable a la hipoxia 3. Este fenómeno es especialmente marcado durante las fases de sueño. El mecanismo fisiopatológico exacto que desencadena la enfermedad permanece desconocido, aunque se han establecido diferentes hipótesis. El hecho de que existan familias con varios miembros afectados, la precocidad con que aparece el cuadro y su asociación con la enfermedad de Hirschsprung en aproximadamente el 20 % de los casos, ha llevado a la presunción de que podría existir un determinante genético 1-3. De hecho, se han asociado diferentes defectos genéticos, entre ellos, mutaciones del gen RET tirosinkinasa, un modulador en el desarrollo de la cresta neural y del sistema parasimpático 4-11; sin embargo, sólo se ha podido demostrar este defecto, u otros, en un número limitado de casos 3.
Desde el año 2003 se ha establecido mediante técnicas de biología molecular la presencia en la gran mayoría de los pacientes afectados de síndrome de Ondine (67-97 % de casos según las series, 0 % de controles) de una mutación en el tercer axón del gen PHOX 2b2,12,13; gen que se encuentra implicado en la diferenciación neuronal durante la embriogénesis. Aunque el mecanismo exacto se desconoce, parece ser que el causante de esta entidad es un trastorno en la integración de la información proveniente de los quimioceptores, probablemente a nivel del tronco del encéfalo 14,15. Por ello, la expresión clínica de la enfermedad se haría más evidente en aquellos momentos en los cuales el resto de mecanismos que regulan la respiración se encuentran inoperantes o menos activos; hecho que acontece durante el sueño, especialmente durante la fase no REM 14,15.
De cualquier manera, el grado de intensidad de la enfermedad entre los pacientes afectados de este síndrome es muy variable; existen enfermos que presentan hipoventilación de forma permanente y otros en los cuales el cuadro se limita a las fases de sueño. Este hecho conduce a que algunos pacientes no sean diagnosticados o se demore el diagnóstico; puede presentarse, entonces, como un cuadro de muerte súbita del lactante o de cor pulmonale secundario a los episodios de hipoxia.
Epidemiología
No existen datos oficiales acerca de la incidencia real de este trastorno, aunque en Francia se cifra la incidencia en 1 caso por 200.000 nacimientos 2. Aunque es muy probable, dado el desconocimiento de este trastorno entre la comunidad médica y la variabilidad de formas clínicas, que se trate de una enfermedad infradiagnosticada 3. Por otro lado, no parecen existir diferencias en cuanto a la distribución por sexo o raza.
Diagnóstico
A pesar de que, asociado a la hipoventilación congénita de intensidad variable, con frecuencia se encuentran asociados hipotonía muscular en grado variable y signos de disfunción del sistema vegetativo, como puedan ser la hipotensión, el déficit de variabilidad de la frecuencia cardíaca, las arritmias (principalmente bradicardia) 16, la sudoración o la distermia basal, etc., no existe ningún signo clínico característico de esta enfermedad. Por ello, el diagnóstico se trata de un diagnóstico de exclusión en un paciente que presenta un cuadro de hipoventilación alveolar. Actualmente los criterios necesarios para el diagnóstico se fundamentan en la existencia de un cuadro de hipoventilación durante el sueño (PaCO2 > 60 mmHg) de inicio durante los primeros meses de vida en ausencia de una enfermedad del tronco del encéfalo, neuromuscular, pulmonar, metabólica o cardíaca que pueda explicar el cuadro 1,3.
El síndrome de Ondine se asocia con relativa frecuencia a afectación de los plexos mioentérico y submucoso del aparato digestivo, con lo cual estos pacientes pueden presentar reflujo gastroesofágico, paresia intestinal y enfermedad de Hirschsprung (20 % de los casos) 1-3. Ocasionalmente existe dificultad para la succión y deglución, probablemente secundaria a la propia disfunción del tronco del encéfalo. Otra asociación frecuente son las anomalías oculares: pupilas mióticas, con frecuencia anisocóricas, con respuesta pobre a la luz (60 % de los casos), estrabismo (50 % de los casos), xeroftalmía por déficit de lágrima, oftalmoplejía 17, etc. Asimismo se ha establecido un aumento del riesgo de desarrollar tumores de la cresta neural (ganglioneuromas, neuroblastomas, ganglioneuroblastomas, etc.) en estos pacientes: 2 % de los casos 1-3.
Actualmente existe la posibilidad de llevar a cabo un diagnóstico molecular mediante técnicas de biología molecular (reacción en cadena de la polimerasa, PCR). Esta técnica, que presenta una adecuada sensibilidad y especificidad, permite detectar una mutación del gen PHOX2b. Asimismo permite dar consejo genético preconcepcional a aquellos padres que planeen tener un nuevo hijo, dado que existe un 10 % de formas mosaico entre los progenitores de estos pacientes 12. Los resultados se obtienen en un plazo de 1-2 semanas. Para su realización tan sólo se requiere una muestra de 3-5 ml de sangre en un recipiente con ácido etilendiaminotetraacético (EDTA) conservada a temperatura ambiente (refrigerada en caso de requerir un transporte de más de 24 h de duración). Cabe subrayar que es posible realizar estudios prenatales mediante muestras obtenidas por amniocentesis.
Diagnóstico diferencial
El diagnóstico diferencial debe excluir todas aquellas entidades que puedan cursar con hipoventilación secundaria o episodios de apnea 1,3:
1. Tumor o malformación del tronco del encéfalo.
2. Ganglioneuroma.
3. Infección del sistema nervioso.
4. Síndrome de Möebius.
5. Enfermedad neuromuscular.
6. Parálisis del diafragma.
7. Miopatía congénita (principalmente miastenia grave congénita).
8. Metabolopatía (enfermedad de Leigh, déficit de piruvato deshidrogenasa, déficit de carnitina, etc.).
9. Cardiopatía congénita.
10. Anomalía estructural de la vía aérea.
11. Sepsis neonatal.
Exploraciones complementarias
Hay que recordar que se trata de una enfermedad cuyo diagnóstico se lleva a cabo tras excluir otras causas 1,3. Por ello, las pruebas irán encaminadas a descartar las entidades referidas en el apartado previo y a valorar la existencia de enfermedades asociadas, como pueda ser el síndrome de Hirschsprung.
En el momento de la sospecha diagnóstica se debe realizar:
1. Analítica sanguínea (hemograma, proteína C reactiva) y hemocultivo.
2. Analítica de líquido cefalorraquídeo (LCR), tinción de Gram y cultivo.
3. Estudio de metabolopatías:
a) Analítica sanguínea: debe incluir aminoacidemia, glucemia, valores de carnitina, amonio, lactato, piruvato y estudio de la piruvato deshidrogenasa.
b) Analítica de orina: debe incluir determinación de aminoácidos, carnitina y ácidos orgánicos.
4. Polisomnografía.
5. Resonancia magnética (RM) craneal y espinal.
6. Ecocardiografía y estudio Holter.
7. Ecografía diafragmática/videofluoroscopia.
8. Fibrobroncoscopia.
9. Si la hipotonía muscular es muy marcada debe valorarse la realización de:
a) Electromiografía y estudios de conducción nerviosa.
b) Biopsia muscular.
Una vez establecido el diagnóstico debe llevarse a cabo:
1. Valoración oftalmológica.
2. Valoración auditiva (potenciales evocados auditivos).
3. Ante la presencia de síntomas digestivos (estreñimiento y distensión abdominal) debe realizarse un enema de colon/biopsia rectal con la finalidad de descartar un síndrome de Hirschsprung.
Tratamiento
El enfoque terapéutico debe ser multidisciplinar, aunque irá dirigido principalmente a mantener una adecuada ventilación.
Soporte ventilatorio domiciliario
Evidentemente las familias requerirán un período de entrenamiento/afianzamiento en el manejo de estos pacientes durante su estancia en las unidades antes de ser remitidos a su domicilio. Los pacientes deben ser monitorizados en el domicilio con pulsioxímetros y un monitor de CO2 espirado. La necesidad de monitorización domiciliaria se hace especialmente necesaria en estos pacientes dado que, debido a la ausencia de respuesta a hipercapnia e hipoxia, pueden no presentar signos clínicos de dificultad ante una situación de compromiso respiratorio 3. La necesidad, en cuanto a horas/día, y la modalidad de este soporte depende del grado de afectación del paciente y, por lo tanto, debe individualizarse según el caso:
Ventilación invasiva
Se plantea como el tratamiento de elección durante los primeros 3-5 años de vida 15,18. Se prefiere el uso de ventiladores ciclados por volumen, y limitados por presión; la modalidad más frecuentemente empleada es la ventilación mecánica intermitente mandatoria sincronizada (SIMV). Es preferible mantener una hiperventilación moderada (PaCO2: 30-35 mmHg) en aquellos pacientes con ventilación mecánica nocturna; puesto que en estos pacientes se ha evidenciado, por polisomnografía, una mejoría en el patrón ventilatorio diurno 1. Evidentemente, el empleo de ventilación mecánica invasiva prolongada requerirá de la realización de una traqueotomía 3.
Ventilación no invasiva
A pesar de que se han utilizado técnicas de ventilación no invasiva con presión negativa 19, parece que actualmente se han sustituido por las técnicas de ventilación no invasiva mediante presión positiva (BiPAP). Aunque se ha descrito casos de lactantes que han sido tratados mediante BiPAP con éxito 20,21, esta técnica ha presentado fracasos frecuentes en pacientes menores de 5 años y representa un mayor riesgo de accidente. Por el contrario, en pacientes mayores de 5 años (principalmente en aquellos que sólo precisan soporte ventilatorio nocturno, en los cuales podría retirarse la traqueostomía) se trata de una excelente opción, con una alta tasa de éxito 1,22. Se utiliza una modalidad asistida/controlada ajustando los valores de IPAP/EPAP según las necesidades del paciente.
Marcapasos diafragmático
El marcapasos diafragmático permite una adecuada ventilación alveolar con una mayor movilidad de la que disponen los pacientes dependientes de un ventilador mecánico 23-25. Por ello, el candidato ideal a recibir esta técnica es aquel paciente mayor de 1-2 años de edad que requiere ventilación mecánica durante 24 h al día y no presenta enfermedad pulmonar 1. Las principales desventajas de esta técnica son el coste, el malestar asociado al marcapasos y la necesidad de reintervenciones quirúrgicas para revisión/recambio del marcapasos.
Medidas generales
1. En pacientes con marcada hipotonía y dificultades para la alimentación debe plantearse la alimentación por sonda nasogástrica para mantener un estado nutricional adecuado. De perpetuarse la situación puede valorarse la necesidad de colocación de una gastrostomía.
2. En pacientes con síntomas destacados de reflujo gastroesofágico o de paresia del tracto digestivo pueden emplearse medidas antirreflujo (normas posturales, leches antirreflujo, etc.) o procinéticos (cisaprida, metoclopramida). Puede ser necesario el empleo de técnicas quirúrgicas antirreflujo.
3. En los pacientes portadores de traqueostomía debe valorarse por el equipo de logopedia la necesidad de colocación de una válvula de Passy-Muir para mejorar su capacidad de comunicación.
Controles
Como ya se ha señalado, el seguimiento debe realizarse de forma multidisciplinar. Por ello, a pesar de que el coordinador del seguimiento debe ser un pediatra neumólogo o un intensivista con experiencia en ventilación mecánica, en el control de estos pacientes intervendrán los servicios de neurología, cardiología, gastroenterología, otorrinolaringología, oftalmología, psicología, trabajo social, logopedia 1,3 (tablas 1-3).
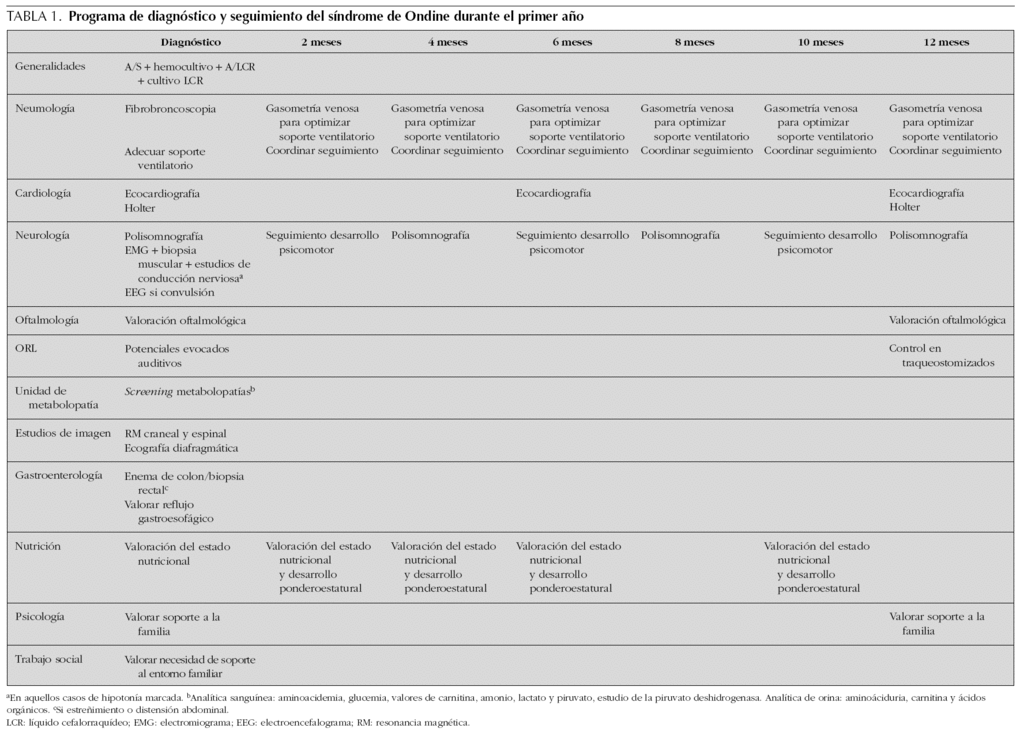


Son especialmente necesarios, como parte de la monitorización del tratamiento, los controles del desarrollo psicomotor y ponderoestatural (en aquellos pacientes con dificultades para la alimentación debe plantearse la alimentación por sonda nasogástrica o la colocación de una gastrostomía para mantener un estado nutricional adecuado; en este caso, debe existir un seguimiento por los servicios de nutrición y cirugía). El pediatra debe visitar al paciente de forma periódica (cada 2-3 meses durante los primeros 3 años y posteriormente de forma anual) con el objetivo de optimizar el soporte ventilatorio; dentro de ello debe incluirse la realización de gasometrías venosas frecuentes (cada 3 meses una vez estabilizado al paciente).
Es recomendable realizar un estudio de polisomnografía cada 4 meses durante los primeros 2 años, cada 6 meses durante el siguiente y anualmente en mayores de 3 años para valorar los cambios en el patrón de respiración conforme se desarrolla el niño.
Al menos anualmente es necesario realizar de forma programada valoraciones por cardiología (es recomendable la realización de una ecocardiografía cada 6 meses y de un estudio Holter anual), oftalmología, neurología (en caso de aparecer crisis comiciales deberá realizarse un electroencefalograma [EEG]).
En pacientes traqueostomizados es especialmente importante un control cada 6 meses por un otorrinolaringólogo. En este grupo de pacientes debe realizarse una broncoscopia cada 24 meses con la finalidad de descartar la presencia de granulomas supraestomales y de hipertrofia adenotonsilar 18.
Evidentemente, los pacientes que presenten síntomas digestivos (enfermedad de Hirschsprung corregida, reflujo gastroesofágico, hipomotilidad intestinal, etc.) deben seguir un control por un gastroenterólogo.
Pronóstico
Conforme existe un mayor conocimiento de la entidad entre la comunidad médica, y a medida que el soporte tecnológico lo ha permitido, ha ido mejorando el pronóstico de estos pacientes 26. De hecho, en el pasado la mayor parte de los pacientes presentaban diferentes grados de retraso psicomotor, retraso del crecimiento, cor pulmonale y episodios comiciales. Secuelas probablemente secundarias, en parte, a los episodios de hipoxemia 3. Actualmente, gracias al diagnóstico precoz y al establecimiento de medidas de soporte ventilatorio, estas secuelas parecen haber disminuido, aunque el porcentaje de retraso psicomotor sigue siendo de alrededor del 25-50 %. Evidentemente, el pronóstico de estos pacientes viene limitado, también, por las complicaciones derivadas de la ventilación mecánica prolongada.
Soporte de otras organizaciones
Las familias pueden encontrar soporte en la organización de familiares afectados de síndrome de Ondine. Para contactar con esta organización puede dirigirse a la dirección www.cchsnetwork.org.
