



El síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS) es una enfermedad hereditaria autosómica dominante que se engloba dentro de los síndromes hereditarios de fiebre periódica, los cuales constituyen a su vez el principal subgrupo dentro de las enfermedades autoinflamatorias sistémicas. El TRAPS se caracteriza por episodios inflamatorios prolongados, recurrentes, en los que se objetiva fiebre y parámetros analíticos inflamatorios elevados que pueden acompañarse de clínica articular, cutánea, ocular y abdominal. Se presenta el caso de un niño de 4 años de edad con episodios inflamatorios recurrentes caracterizados por fiebre asociada a manifestaciones cutáneas diagnosticado de TRAPS. Se revisan los hallazgos clínicos, analíticos, diagnóstico genético y tratamiento de esta enfermedad.
Tumoral necrosis factor receptor-associated periodic syndrome (TRAPS) is an autosomal dominantly inherited disease belonging to the hereditary periodic fever syndromes, which are the main subgroup among systemic autoinflammatory diseases. TRAPS is characterized by prolonged and recurrent inflammatory attacks associated with fever and an acute phase reaction. Articular, cutaneous, ocular and abdominal symptoms may also be present. We describe the case of a 4-year-old boy with recurrent inflammatory episodes, fever and cutaneous symptoms who was diagnosed with TRAPS. We review the clinical and laboratory findings, genetic diagnosis, and treatment approach in this disease.
En el año 1982 se describió una entidad hereditaria nueva, en una familia irlandesa, denominada fiebre hiberniana familiar (FHF), que presentaba un patrón de herencia autosómico dominante y se caracterizaba por la presencia de episodios febriles prolongados y recurrentes que podían asociarse a mialgias y manifestaciones cutáneas migratorias1. Posteriormente se describieron otras familias afectadas del mismo síndrome con orígenes étnicos diversos2–4. En el año 1999 se describió el defecto molecular de estas entidades al identificarse mutaciones en el receptor 1 del factor de necrosis tumoral alfa (TNF-α), también denominado p55 y CD120a, y se acuñó el término de síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS) para designar todos los casos, con independencia de su origen étnico5.
El TRAPS se engloba dentro de los síndromes hereditarios de fiebre periódica, que a su vez representan el subgrupo más numeroso dentro de las enfermedades autoinflamatorias sistémicas6,7. Estas enfermedades se caracterizan por episodios inflamatorios recurrentes, sistémicos, en ausencia de etiología infecciosa o autoinmune, y que se deben a defectos en la regulación del proceso inflamatorio5 (tabla 1). Todas estas enfermedades suelen iniciarse en la edad pediátrica, por lo que un diagnóstico de sospecha de estas patologías por parte del pediatra podría evitar numerosos exámenes complementarios y tratamientos inadecuados, así como un abordaje terapéutico óptimo y precoz y una correcta valoración del pronóstico en cada caso.
Clasificación de las enfermedades autoinflamatorias sistémicas hereditarias
|
CINCA: síndrome crónico, infantil, neurológico, cutáneo y articular; CRMO: osteomielitis crónica multifocal recurrente; HIDS: síndrome de hiper-IgD asociado a fiebre periódica; NOMID: enfermedad neonatal multisistémica inflamatoria; PAPA: artritis piogénica estéril, pioderma gangrenoso y acné; TRAPS: síndrome periódico asociado al receptor del factor de necrosis tumoral.
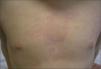
Paciente de 4 años presenta fiebre elevada de 11 días de evolución acompañada de diarrea y un exantema eritematoso troncular de 24h de evolución. Como antecedentes patológicos destacan tres episodios de fiebre prolongada asociada, en dos ocasiones, a un exantema cutáneo en el primer, segundo y al tercer año de vida. El segundo episodio tuvo lugar tras el tratamiento quirúrgico de una fractura de fémur. En el examen físico, el paciente presenta un exantema eritematoso maculopapular de morfología anular que desaparece a la vitropresión y de distribución troncular (fig. 1), inyección conjuntival y edema periorbitario bilateral. Durante el ingreso la fiebre se prolongó 7 días, con aparición intermitente del exantema y disminución progresiva de los parámetros analíticos inflamatorios hasta su normalización. En las exploraciones complementarias destacaba: leucocitos 21.700/μl (neutrófilos, 78 %; cayados, 9 %; linfocitos, 10 %; monocitos, 3 %), proteína C reactiva (PCR) 277mg/l, procalcitonina cualitativa > 10ng/dl. La dosificación de inmunoglobulinas fue normal (IgG, 12.340mg/l, IgA, 1.258mg/l; IgM, 1.765mg/l; IgD, 32 UI/ml). Las serologías para citomegalovirus, rubéola, virus de Epstein-Barr, virus de la inmunodeficiencia humana, virus de la hepatitis B y C y parvovirus B19 fueron negativas. El sedimento urinario, el urocultivo, los coprocultivos y los hemocultivos seriados fueron negativos. Se realizaron radiografías de tórax y abdomen y un ecocardiograma sin hallazgos patológicos. Dados los antecedentes patológicos y las características del cuadro, se solicitó el estudio genético de enfermedades autoinflamatorias sistémicas hereditarias, que demostró la presencia de la variante genética Cys-30-Arg (C30R) en heterocigosis en el exón 2 del gen TNFRSF1A, el cual codifica para el receptor 1 del TNF. Dicha variante ha sido descrita previamente en la bibliografía médica asociada a enfermedad en pacientes afectados de TRAPS5. El estudio genético realizado a los padres fue negativo. Se instauró tratamiento puntual con corticoides orales durante las reagudizaciones, con buena respuesta clínica y analítica, sin precisar tratamiento en los períodos intercrisis. El paciente realiza controles periódicos de concentraciones de velocidad de sedimentación globular (VSG), PCR y proteína sérica amiloide A.
DISCUSIÓN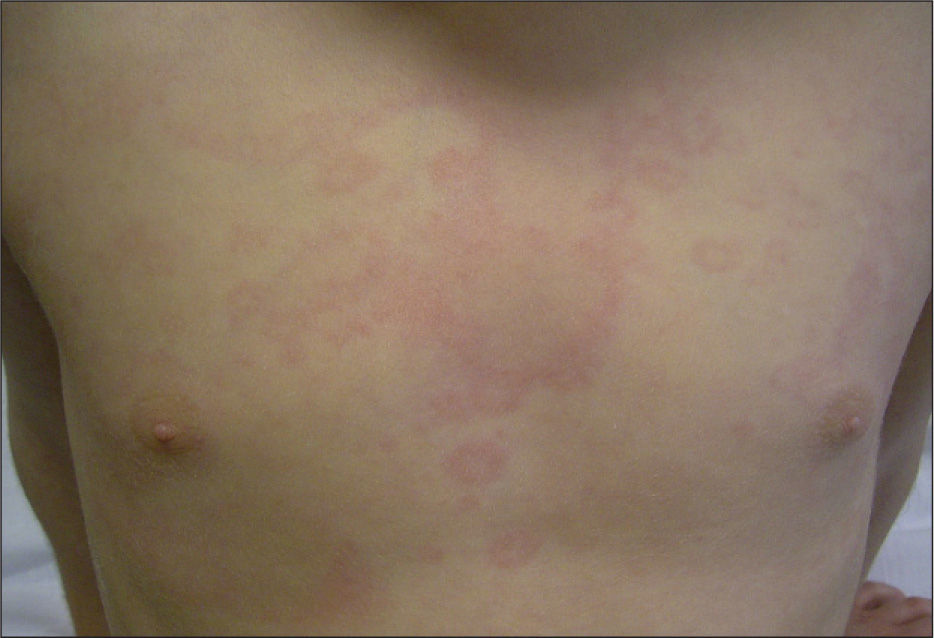
Este caso corresponde a una mutación de novo para el gen del receptor 1 del TNF-α ya que el estudio genético de los progenitores demostró que ninguno de ellos era portador de la mutación. El gen del receptor del TNF-α se localiza en el brazo corto del cromosoma 12, y hasta el momento se han descrito más de 50 mutaciones responsables del síndrome8. La mutación de este gen condiciona una pérdida del control sobre las acciones biológicas del TNF-α, prolongando sus efectos en tiempo e intensidad. El inicio de la enfermedad se sitúa por debajo de los 10 años de vida, pero de forma retrospectiva es posible identificar manifestaciones propias de la enfermedad a edades más tempranas9. La frecuencia e intensidad de las crisis son variables y, como en este caso, suelen asociarse a factores precipitantes como infecciones, traumatismos, menstruación y estrés9. Entre las manifestaciones clínicas, la fiebre es un signo casi constante; suele ser elevada y puede mantenerse durante días o semanas hasta disminuir progresivamente. Los dolores abdominales de tipo cólico son el segundo síntoma en frecuencia y pueden confundirse con un abdomen quirúrgico, lo cual puede deberse tanto a una peritonitis inflamatoria aséptica como a una inflamación de los músculos de la pared abdominal. Las manifestaciones cutáneas ocupan el tercer lugar y son muy variables, desde máculas eritematosas migratorias o placas de seudocelulitis muy sugestivas de este síndrome hasta lesiones purpúricas, que son menos frecuentes9. El estudio histológico de las lesiones se caracteriza por un infiltrado superficial y profundo, perivascular e intersticial linfomonocitario, sin evidencia de formación de granulomas o vasculitis. También es habitual la aparición de mialgias migratorias que suelen asociarse a un determinado grupo muscular, rigidez articular, artralgias y, con menos frecuencia, verdaderas artritis asépticas que se resuelven sin secuelas. Las adenopatías pueden presentar una distribución difusa o estar localizadas en la región cervical. La diarrea, el dolor testicular, el edema periorbitario y la conjuntivitis aséptica bilateral son síntomas que también se han descrito durante las crisis de esta enfermedad9.
Las pruebas complementarias realizadas durante una crisis revelarán un aumento de los parámetros inflamatorios (PCR, VSG, procalcitonina y fibrinógeno), leucocitosis con neutrofilia, trombocitosis y anemia de enfermedades crónicas en el caso de crisis prolongadas. No se encuentran autoanticuerpos, la concentración de inmunoglobulinas es normal o discretamente elevada (sobre todo la IgA) y las pruebas microbiológicas son negativas. La confirmación diagnóstica se realiza mediante el estudio genético; es importante efectuar este estudio genético a los familiares a fin de establecer el carácter de novo o no de la mutación y detectar la posible presencia de portadores asintomáticos de la mutación.
El diagnóstico diferencial debe realizarse fundamentalmente con otras enfermedades autoinflamatorias sistémicas y, especialmente, con los otros síndromes hereditarios de fiebre periódica, que cursan con fiebres recurrentes y/o persistentes, sobre todo si existen antecedentes familiares (v. tabla 1).
En el tratamiento del TRAPS, los antiinflamatorios no esteroideos son parcialmente eficaces sobre ciertos síntomas como la fiebre, las artralgias y las mialgias, pero no son capaces de disminuir ni la duración ni la frecuencia de los episodios. Los corticoides pueden reducir la duración de las crisis pero no su frecuencia, y con el tiempo se ha descrito tolerancia a su acción, lo que obliga a aumentar progresivamente la dosis para obtener el mismo efecto10. Dado el mecanismo patogénico subyacente en el TRAPS, se ha postulado el tratamiento con fármacos anti-TNF, y en particular con etarnecept, que está consituido por dos receptores p75 del TNF asociados al fragmento Fc de la IgG19. Los estudios realizados hasta el momento son escasos y con un número de pacientes limitado, si bien parecen sugerir una disminución de los síntomas y de las dosis de corticoides necesarias, sin una clara correlación con la disminución de los parámetros inflamatorios9–11. Recientemente se ha dado a conocer una excelente respuesta clínica y bioquímica en un paciente afectado de TRAPS y tratado con el bloqueador de la interleucina-1 anakinra12. El pronóstico de vida está determinado principalmente por la posible aparición en el curso de la enfermedad de amiloidosis del tipo AA. Esta complicación ocurre en un porcentaje variable de los casos según las series (2–25 %) y es más frecuente en mutaciones en las que está implicado un residuo de cisteína9.
Dado que las crisis febriles son espaciadas y no existen parámetros inflamatorios persistentes durante los intervalos asintomáticos, en este caso se ha optado por administrar tratamiento puntual con corticoides durante los episodios febriles. Al paciente se le realiza un seguimiento clínico y analítico durante los brotes y en períodos intercrisis, incluyendo controles periódicos de proteína sérica amiloide A ya que el paciente es portador de una mutación que se relaciona con un aumento del riesgo para desarrollar amiloidosis. Debido a la buena respuesta, de momento no se ha instaurado tratamiento con fármacos anti-TNF.
En resumen, se trata de un paciente con episodios febriles prolongados que se acompañan de lesiones cutáneas y de un cuadro inflamatorio sistémico, diagnosticado de TRAPS. El conocimiento de esta entidad puede permitir el diagnóstico precoz de estos pacientes, lo que facilita su tratamiento.
