



El síndrome de Sjögren-Larsson (OMIM #270200) es un trastorno neurocutáneo caracterizado por ictiosis congénita, discapacidad intelectual y espasticidad1. Está causado por mutaciones en el gen ALDH3A2, codificante para la enzima aldehído graso deshidrogenasa, implicada en la oxidación de alcoholes en ácidos grasos2,3.
Se describe la variabilidad clínica y genética de 3 casos diagnosticados en nuestro servicio.
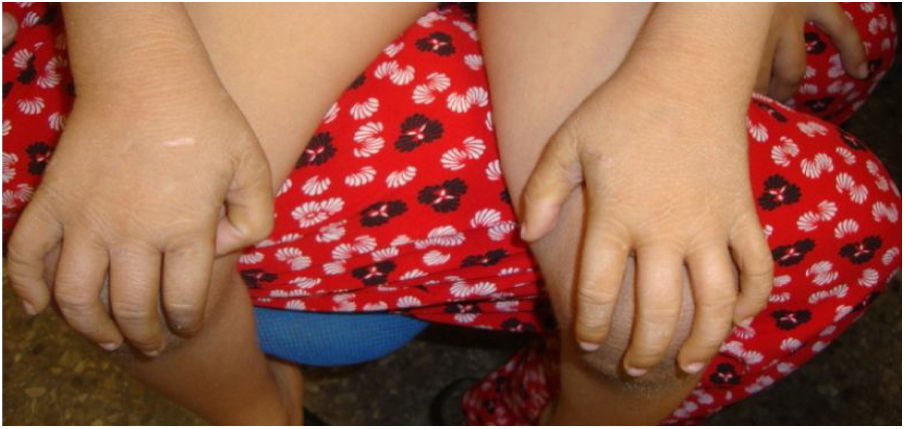
El caso 1 es la primera hija de una pareja no consanguínea y sin antecedentes a destacar. El parto se produce a las 36 semanas de gestación, objetivándose una leve eritrodermia que evoluciona a una ictiosis vulgar (según la descripción de la biopsia cutánea) (fig. 1).
Es remitida a Neuropediatría a los 17 meses por hipotonía axial severa, con sedestación inestable e ictiosis. Los estudios metabólicos, neurofisiológicos y de neuroimagen hechos hasta entonces fueron normales.
Aunque no presentaba otros signos de ictiosis sindrómica4, se realizó un estudio de hibridación genómica comparada, donde se detectó una deleción de 159,6kb en la región 17p11.2, hg19 (chr17:19424245-19583843), que incluía el gen ALDH3A2. La secuenciación de la otra copia del gen mostró una mutación con desplazamiento de la pauta de lectura en el exón 1 (c.86_96del, p.Ala29Aspfs*21), que da lugar a una proteína truncada.
El estudio de segregación familiar confirmó que ambas alteraciones son heredadas (una de cada progenitor).
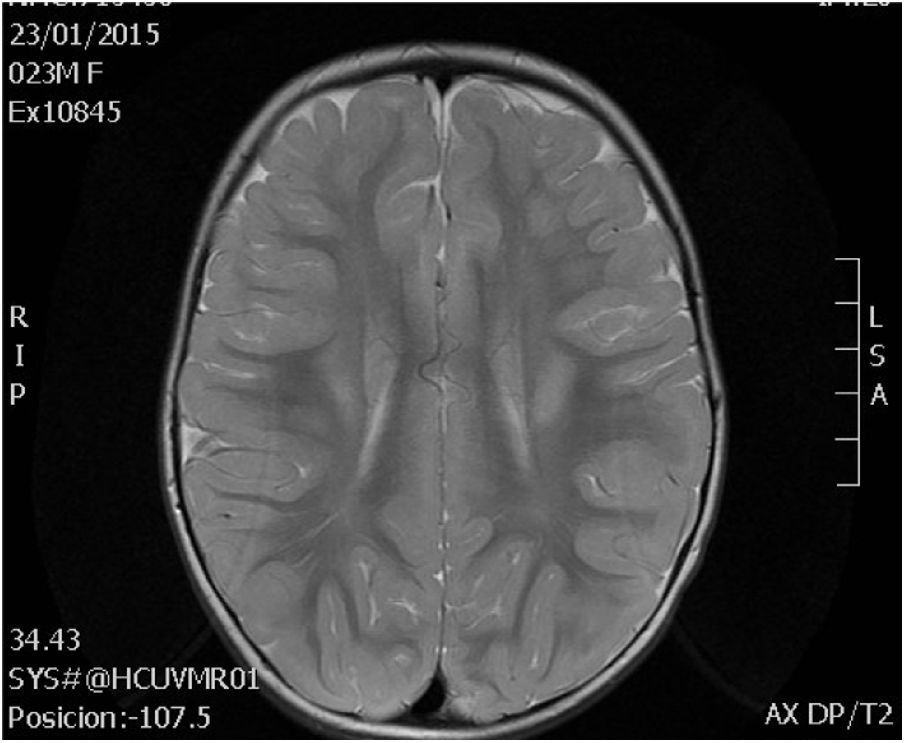
La resonancia magnética cerebral a los 2 años mostró alteración en la mielinización (fig. 2). Actualmente, a los 6 años, presenta discapacidad intelectual leve e hipotonía axial con espasticidad de miembros inferiores, que imposibilita el desplazamiento autónomo.
Ha tenido 5 convulsiones febriles típicas, sin precisar tratamiento antiepiléptico. No presenta alteraciones oftalmológicas en la actualidad.
Los casos 2 y 3 son hermanas en una familia sin consanguinidad referida. Al nacimiento a las 34 y 36 semanas de gestación, respectivamente, ambas presentaron ictiosis moderada, descrita como lamelar. Fueron capaces de mantener sedestación sobre los 9 meses y deambulación autónoma entre los 4 y 6 años. La paciente 2 aún mantiene deambulación autónoma a los 22 años. Ambas desarrollaron signos de espasticidad durante los 2 primeros años de edad, y tras la obtención de neuroimagen con alteración de la mielinización, se sospechó el diagnóstico.
Ambas presentaron convulsiones febriles de características típicas, y asocian retraso del lenguaje y discapacidad intelectual leve.
A los 14 y 16 años, respectivamente, se diagnosticaron inclusiones en el fondo de ojo.
El diagnóstico se realizó mediante actividad enzimática en fibroblastos y estudio molecular del gen ALDH3A2. Ambas pacientes presentan la variante ya descrita c.471+1delG (NM_000382.2) en homocigosis.
El estudio de segregación familiar confirmó que ambos progenitores son portadores de la mutación.
En junio de 2020 aparecen publicadas 280 variantes del gen ALDH3A2 en 182 individuos con síndrome de Sjögren-Larsson (LOVD, PubMed). Algunas de ellas, como c.471+1delG, presente en las pacientes 2 y 3, han sido descritas ampliamente en familias europeas. Sin embargo, los cambios detectados en la primera paciente no han sido descritos previamente5.
Tras una revisión de los casos publicados en nuestro país, encontramos 4 diagnosticados en la edad adulta, tras el diagnóstico de la enfermedad oftalmológica o dermatológica características, y un solo caso de presentación pediátrica6.
La mutación c.86_96del da lugar a un desplazamiento en la pauta de lectura y una proteína truncada. Sin embargo, la presencia de un codón de inicio de lectura más adelante podría hipotetizar la presencia de una proteína en la que falten los primeros 32 aminoácidos (de los 508 que contiene la proteína), y que pueda mantener cierta actividad residual y un fenotipo atenuado.
Pensamos que es importante recordar la asociación de ictiosis congénita y enfermedad neurológica como uno de los síndromes neuroictiósicos conocidos, así como llamar la atención sobre la presencia de portadores de estas mutaciones en nuestro país, para ayudar a un diagnóstico precoz y al establecimiento de una adecuada correlación fenotipo/genotipo. El caso 1, que presenta una deleción y una mutación no descrita, tiene un patrón con mayor hipotonía axial y con una ictiosis más leve que los casos 2 y 3, que muestran una mutación ya descrita ampliamente.
