





La utilización en el pasado de una terminología imprecisa para designar a los tumores vasculares infantiles ha contribuido durante años a diagnósticos incorrectos y, como consecuencia, a tratamientos inadecuados.
En la infancia pueden presentarse diferentes tipos de tumores vasculares, como los hemangiomas infantiles, que son con diferencia los más frecuentes, y otros mucho más raros, como los hemangiomas congénitos (rápidamente involutivo y no involutivo), el hemangioendotelioma kaposiforme, el angioblastoma o angioma en penacho, o el granuloma piógeno. Su correcto conocimiento y diagnóstico, siempre en el contexto de un equipo multidisciplinario, es imprescindible para reducir errores diagnósticos, exámenes complementarios y pruebas invasivas innecesarias, y así, si fuera preciso, recibir el tratamiento más indicado y efectivo en cada caso.
En el presente artículo revisamos la evolución histórica en cuanto a la nomenclatura y clasificación de las lesiones vasculares, las diferentes características clinicopatológicas de cada uno de los tumores vasculares, los exámenes complementarios indicados para llegar a un correcto diagnóstico, su diagnóstico diferencial y los distintos tipos de tratamiento que existen con sus indicaciones más reconocidas, en el momento actual, para los diferentes tumores vasculares y situaciones clínicas concretas.
The use in the past of an imprecise terminology to designate vascular tumors has contributed to its incorrect diagnosis, and as a consequence, to inadequate treatment.
In childhood, different types of vascular tumors may be present. Hemangiomas of infancy are by far the most frequent, and other less common types are congenital hemangiomas (rapidly involuting or RICH and non-involuting or NICH), kaposiform hemangioendothelioma, angioblastoma or tufted angioma and pyogenic granuloma. The correct knowledge and diagnosis, always in a multidisciplinary setting, is required to reduce incorrect diagnosis, unnecessary complementary examinations and invasive tests, and for the patient to receive the most effective and precise treatment in each case.
This article reviews the historical evolution, nomenclature and classification of vascular lesions, the different clinical and pathological characteristics of each vascular tumor, the complementary examinations required correct diagnosis, the differential diagnosis, as well as highlighting the treatment options currently available for different vascular tumors and related clinical conditions.
El estudio de la angiogénesis y la necesidad de encontrar tratamientos que inhiban la formación de vasos sanguíneos en los tumores malignos han dado paso en los últimos 20 años a una revolución conceptual y nosológica en el conocimiento de los tumores vasculares1–7. Desgraciadamente, estos avances no han llegado con la fluidez necesaria a la práctica clínica. Prueba de esto es que, en un reciente estudio de uno de los centros pediátricos más prestigiosos del mundo y el de mayor experiencia en el tratamiento de diferentes anomalías vasculares infantiles (Children′s Hospital de Boston), el 75% de los pacientes allí remitidos tenía un diagnóstico y un tratamiento previos incorrectos, y del 25% diagnosticados correctamente, un 14% había recibido un tratamiento inadecuado6. Estos datos son el fiel reflejo de la difícil realidad que afrontan los niños con anomalías o lesiones vasculares (tumores y malformaciones vasculares) y son totalmente extrapolables a la experiencia que ocurre en nuestros hospitales. Por tanto, parece necesario, además de una correcta clasificación nosológica de las anomalías vasculares, un enfoque multidisciplinario (pediatras, dermatólogos, cirujanos pediátricos, plásticos y vasculares, angiorradiólogos, otorrinolaringólogos, oftalmólogos y anatomopatólogos) respecto al seguimiento y las posibilidades terapéuticas de muchos de estos pacientes2,3,7,8.
En la actualidad, con un correcto diagnóstico de las diferentes lesiones vasculares, entre las que se incluyen los tumores vasculares, y en el contexto de un equipo multidisciplinario se pueden obtener resultados terapéuticos efectivos, disminuir los errores diagnósticos, evitar las consultas a diferentes especialistas en función de la localización (“pacientes nómadas”), reducir los exámenes complementarios, en ocasiones invasivos, y conseguir la escolarización-socialización del niño con las mínimas secuelas psicológicas3,6,7.
Clasificación de las lesiones vascularesLa nomenclatura, sin duda, ha sido el mayor obstáculo para el conocimiento de los tumores vasculares en la infancia. Hasta hace unos pocos años, los tumores vasculares infantiles se conocían genéricamente como “angiomas” (planos, cavernosos, etc.), “hemangioendoteliomas” (HE), “linfangiomas” o “hemolinfangiomas”, y aludían a una clasificación puramente descriptiva. En 1982, John Mulliken y Julia Glowacki publicaron su trabajo “Hemangiomas y malformaciones vasculares en la infancia: clasificación basada en las características endoteliales” y pusieron fin a décadas de confusión terminológica distinguiendo taxativamente los tumores de las malformaciones vasculares1,9.
En 1992 se funda la International Society for the Study of Vascular Anomalies con el objeto de aglutinar y consensuar a profesionales de diversos campos de la medicina que están en contacto con estos pacientes y así mejorar el conocimiento de la etiopatogenia, el diagnóstico y el tratamiento de los pacientes con lesiones vasculares. En 1996 edita la clasificación que actualmente sigue vigente (tabla 1). Queda claro desde entonces que lo que antes se denominaba “angioma en fresa” es un hemangioma infantil (HI), un “angioma plano” es realmente una malformación capilar o un “angioma cavernoso” es una malformación venosa. De forma simultánea, se describen nuevos tumores vasculares diferentes del HI, como los hemangiomas congénitos (HC) rápidamente involutivos (RICH rapidly involuting congenital hemangioma), los HC no involutivos (NICH non involuting congenital hemangioma) o el HE kaposiforme (HEK)10–14.
Clasificación de las lesiones vasculares (adaptada de la International Society for the Study of Vascular Anomalies Classification)7
| Tumores vasculares: |
|
| Malformaciones vasculares: |
|
HEK: hemangioendotelioma kaposiforme; NICH: hemangioma congénito no involutivo; RICH: hemangioma congénito rápidamente involutivo.
En 2001 North descubre el marcador inmunohistoquímico GLUT-1 que identifica y separa definitivamente el hemangioma del resto de los tumores. Otros marcadores celulares (D2-40, Proxy-1, Ephrin B, etc.) también han ayudado a la distinción entre diferentes lesiones vasculares7,8,15–17.
En 2004, los grupos de Boston y Arkansas clasifican los tumores vasculares hepáticos infantiles y rechazan definitivamente su denominación como “HE hepáticos”. Conviene recordar que el hemangioma es un tumor exclusivamente pediátrico, por lo que la denominación de “angiomas hepáticos, esplénicos o vertebrales” en el adulto es del todo errónea. Estas lesiones, en otras edades, son sistemáticamente malformaciones venosas y su evolución clínica difiere radicalmente de los hemangiomas hepáticos8,18–20.
Hemangiomas infantilesSon los tumores más frecuentes en la infancia. Algunos estudios refieren una incidencia del 5% en todos los niños de raza blanca, hasta un 22% de los prematuros con menos de 1.000 g al nacimiento, y una menor incidencia en grupos étnicos no caucásicos.
Son 4 veces más frecuentes en el sexo femenino, especialmente los segmentarios3,7,21–25.
EtiopatogeniaAunque la presencia de HI es frecuente en hermanos, la mayoría son esporádicos sin implicación de factores hereditarios, excepto en casos excepcionales de HI familiares con un posible patrón de herencia autosómico dominante7,26,27.
En cuanto a su origen, 2 teorías permanecen vigentes sometidas a constante investigación: 1) origen trofoblástico o placentario (similitud celular, inmunológica y molecular de hemangioma y placenta; mujeres que se les realizaron biopsias coriales tienen mayor tasa de hijos con HI), y 2) teoría de la vasculogénesis (proceso por el que las células precursoras del endotelio originan vasos sanguíneos) y la angiogénesis (desarrollo de nuevos vasos a partir de los existentes)7,8,28–31.
Aunque se desconoce la fisiopatología exacta del crecimiento y de la involución de los HI, parece que durante la fase proliferativa se forman acúmulos densos de células endoteliales que forman pequeños capilares. Mediante técnicas inmunohistoquímicas se han detectado marcadores celulares, incluyendo antígeno nuclear de proliferación celular, colagenasa tipo iv, factor de crecimiento fibroblástico básico (bFGF), urocinasa y selectina-E. Además, se ha indicado la implicación de factores hormonales y sustancias angiogénicas segregadas por diversos tipos celulares7,32. Todos los HI presentan inmunorreactividad histológica positiva frente al “GLUT-1”, lo que permite distinguirlos de los HC y de las malformaciones vasculares7,8,15,16. La importancia de la vasculogénesis es más controvertida, aunque la presencia de arterias anómalas en algunos hemangiomas extensos se ha atribuido a defectos del desarrollo que se producen alrededor de la semana 8 o 10 de la gestación. Los mecanismos causantes de la involución espontánea son aún más desconocidos16,27,33,34.
Características clínicasSe caracterizan por ser de aparición y crecimiento posnatal (aunque el 30–50% pueden estar presentes al nacimiento), y pueden proliferar durante los primeros meses de vida para involucionar siempre antes de la pubertad. Por tanto, es un tumor exclusivo de la infancia y no existe en la vida adulta. Se localizan más frecuentemente en la cabeza y en el cuello (80%), seguido del tronco y de las extremidades, aunque pueden situarse en cualquier parte del cuerpo1–4,7,22–25,29,35–37.
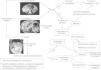
Todo HI sigue una evolución característica en el tiempo (fig. 1), y si no es así, no es ni debe llamarse hemangioma3,4,7,22,38,39:
- •
Fase inicial: al principio se manifiestan como zonas pálidas, equimóticas, telangiectasias, máculas rosadas, micropápulas rojizas agrupadas o úlceras (lesión precursora). El diagnóstico diferencial en este momento puede ser muy amplio.
- •
Fase proliferativa: entre la segunda y la sexta semana de vida aumenta de tamaño, y se transforman en pápulas, placas, nódulos o tumores de color rojo intenso y superficie lisa o lobulada. Durante esta fase (primeros 6 o 18 meses) pueden duplicar o triplicar su tamaño. Posteriormente estabilizan su tamaño.
- •
Fase involutiva: los signos indicativos de regresión son la aparición de tractos fibrosos blanquecinos en su superficie y de una coloración gris-rosada o violácea. Un 50% de las lesiones ha involucionado completamente a los 5 años, un 75% a los 7 años y un 90% a los 9 años. Cuanto antes aparecen los signos de regresión, mejor y más temprano es el resultado estético final. Los hemangiomas localizados en la punta nasal, los labios y la zona parotídea involucionan con más lentitud. En general, los que más tardan en involucionar suelen dar lugar a una lesión residual proporcional al tamaño inicial.
El HI cutáneo según la localización de la proliferación vascular puede ser superficial, profundo y compuesto o mixto, depende de la localización histológica en la piel y del color de la lesión, según su profundidad2,3,6–8,22–24,29:
- •
HI superficiales (62%): son los mal llamados “angiomas capilares”. Se ubican en la parte superficial de la dermis, son tumores lobulados, de color rojo intenso o violáceos, bien delimitados, con capilares diminutos que protruyen en su superficie y les confieren su morfología típica “en fresa”. El tamaño es variable, la consistencia es blanda y no desaparecen completamente con la vitropresión.
- •
HI profundos: son los mal llamados “angiomas cavernosos”. Se localizan en la dermis profunda y en la hipodermis. Son menos frecuentes (15%) y se manifiestan como placas, nódulos o tumores eritematoazulados (a veces el color de la piel es normal), están mal delimitados. La superficie puede ser irregular, con venas dilatadas y telangiectasias.
- •
HI compuestos o mixtos (22%): coexisten los componentes superficial y profundo.
Habitualmente, los HI se presentan como una lesión cutánea única, pero en un 5% de los casos las lesiones son múltiples. Cuando hay 5 o más hemangiomas (hemangiomatosis neonatal), hay mayor probabilidad de presentar hemangiomas en órganos internos3,7,8,18.
En cuanto a su distribución pueden ser de la siguiente manera7,13,29,37,39:
- •
HI focales: son lesiones redondeadas, que pueden trazarse con un compás, como nódulos o placas. Generalmente son asintomáticos.
- •
HI segmentarios: se presentan como placas lineales o con un patrón geográfico (desarrollo de metámeras). Son más propensos a asociar complicaciones, tales como ulceración, malformaciones asociadas y situaciones de riesgo vital.
a. Complicaciones relacionadas con el propio hemangioma:
- •
Ulceración: es la complicación más frecuente (5–10%). Se debe a la isquemia y necrosis de la lesión. Es más frecuente en los segmentarios (29%) que en los focales (8%). Suele ocurrir durante la fase de crecimiento rápido y se relaciona directamente con su tamaño y profundidad. Afecta sobre todo a los hemangiomas localizados en la cabeza y en el cuello, así como en la zona periorificial (especialmente labios, perioral o anogenital). Estas lesiones son muy dolorosas, provocan irritabilidad e insomnio y, según la localización, dificultan la ingesta, la defecación o la micción2,3,8,23–25. En la patogénesis parecen implicados factores como trauma local, infecciones bacterianas e hipoxia7,8,40. En general, las lesiones ulceradas tienden a involucionar más rápidamente. Los hemangiomas ulcerados tienen el riesgo de sobreinfectarse, con destrucción de los tejidos blandos o del cartílago subyacente, y de formar cicatrices que obligan a un tratamiento quirúrgico posterior3,7,40–41.
- •
Hemorragia: las hemorragias profusas son complicaciones infrecuentes de los hemangiomas cutáneos, aunque sí es común una hemorragia leve o moderada en los ulcerados, que suelen resolverse mediante compresión directa7,39–40.
- •
Insuficiencia cardíaca: en hemangiomas de gran tamaño, sobre todo si existen hemangiomas hepáticos asociados, puede producirse una insuficiencia cardíaca congestiva (ICC) por alto gasto8,18,42.
- •
Secuelas estéticas: a pesar de su carácter involutivo, entre un 20 y un 40% de los hemangiomas deja cambios cutáneos residuales, como telangiectasias, hipopigmentación, cicatrices atróficas y masas fibroadiposas redundantes. Éstas son más importantes en las lesiones dérmicas de gran tamaño, las de mayor profundidad, en localizaciones como nariz-labios-pabellón auricular, y en los que se ulceran2,3,7,24,40,43.
- 1c?>
b. Complicaciones relacionadas con la localización:
La localización del hemangioma es un factor determinante de su gravedad. Las lesiones pequeñas que proliferan lentamente pueden ser muy problemáticas, incluso pueden suponer riesgo vital si afectan a estructuras anatómicas y funcionalmente importantes7,8.
- •
Hemangioma periorbitario: especialmente los localizados en el párpado superior suponen un riesgo para la visión y deben valorarse cuidadosamente. La complicación más frecuente es el astigmatismo, secundario a la compresión y la deformidad corneal. Si las lesiones impiden una correcta apertura palpebral, pueden provocar ambliopía. Dado que se trata de una alteración irreversible al cabo de unos pocos días, cuando afecta a niños menores de un año, se considera una verdadera urgencia oftalmológica. Estos hemangiomas orbitopalpebrales también pueden invadir el espacio retrobulbar, comprimir el nervio óptico y ocasionar proptosis y ceguera35–37,44–46.
- •
Hemangioma de la vía aérea superior: tradicionalmente llamados hemangiomas subglóticos. Pueden producir obstrucción de la vía aérea. El 85% son focales y el 15% son segmentarios. No obstante, se estima que sólo un 10–20% de los hemangiomas subglóticos son sintomáticos antes de involucionar espontáneamente. Los síntomas suelen aparecer entre las 6 y las 12 semanas de vida, incluyendo crisis de tos seca, estridor inspiratorio, ronquera, cianosis, tiraje, disnea e insuficiencia respiratoria aguda. La mitad de estos pacientes tiene hemangiomas en otras localizaciones. Los hemangiomas cutáneos que tienen un riesgo más elevado de extenderse hacia la vía aérea y provocar sintomatología son los localizados en la región preauricular, mentón, cervical anterior y labio inferior (región mandibular)7,35,36,47,48. Los hemangiomas que afectan las fosas nasales también pueden provocar sintomatología respiratoria en el período neonatal, sobre todo durante la ingesta35,37.
- •
Hemangioma periauricular y parotídeo: pueden ocluir el conducto auditivo externo y provocar una hipoacusia de transmisión reversible cuando involucionan. Excepcionalmente, las lesiones de gran tamaño pueden ocasionar hipertrofia de los huesos mandibulares, maxilares y del pabellón auricular, por incremento del flujo sanguíneo local7,41,49. Suelen precisar tratamiento prolongado con corticoides orales (6–9 meses). La cirugía tiene un papel limitado dada la alta vascularización, la dificultad de preservar el nervio facial y el requerimiento de embolización preoperatoria8,41,45,49,50.
- •
Hemangioma gastrointestinal: puede producir hemorragias digestivas, habitualmente bajas51.
- •
Hemangioma hepático: probablemente conforma el grupo de diagnóstico y tratamiento más complejo. En la actualidad hay consenso en el seno de la International Society for the Study of Vascular Anomalies de que los tumores vasculares hepáticos no difieren de los cutáneos en su patrón inmunohistoquímico, y comparten los mismos marcadores celulares, por lo que deben denominarse de la misma manera. La diferencia de comportamiento se enmarca en el contexto de las diferencias entre el hígado y la piel como órgano diana8,18,20,42.
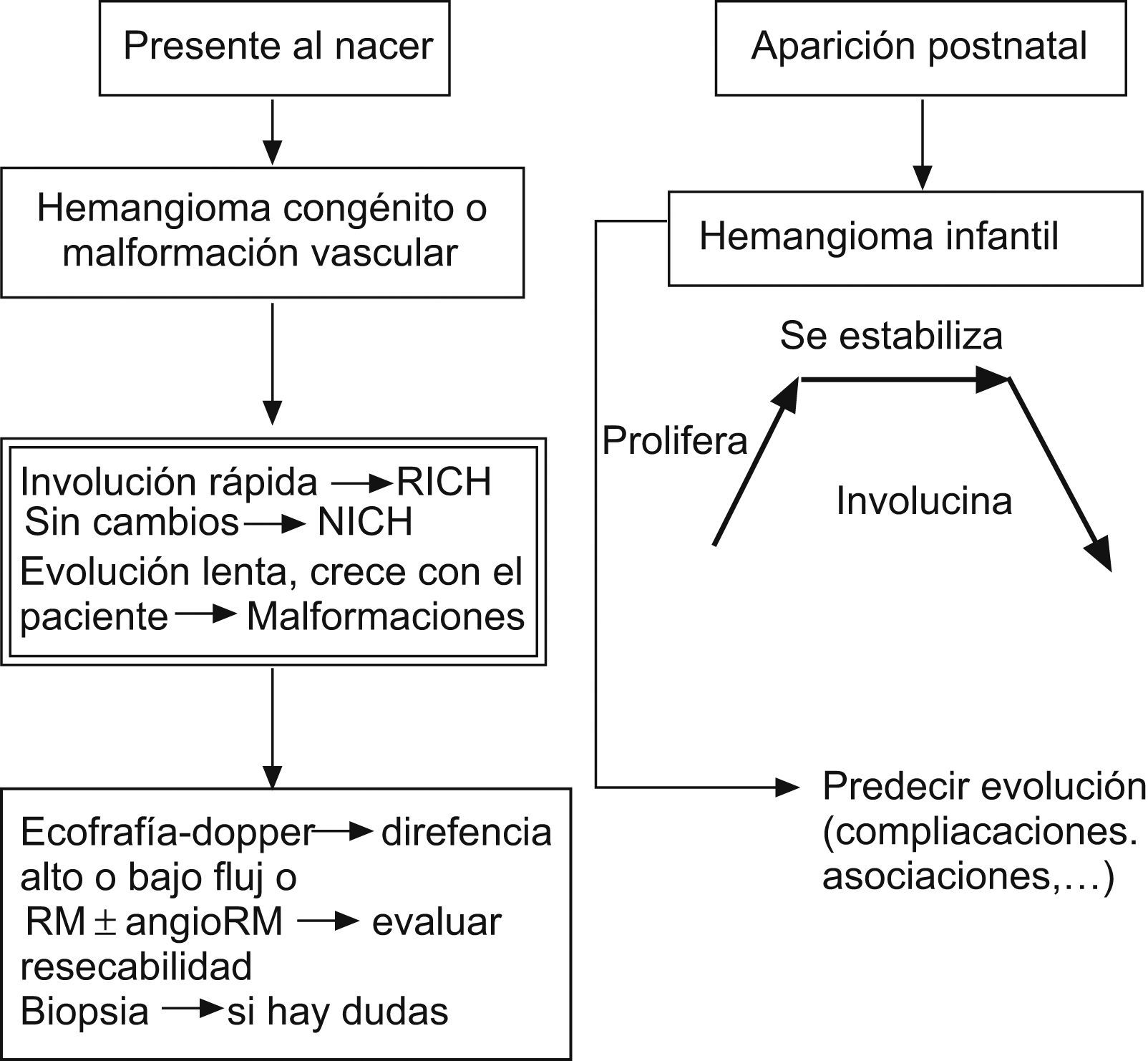
Tipos de tumores vasculares hepáticos en la infancia (fig. 2):
- 1.
Hemangioma hepático solitario: corresponde a un RICH, por lo tanto, es GLUT-1 negativo. Son grandes lesiones presentes al nacimiento, a menudo diagnosticadas prenatalmente. No prolifera durante los primeros meses de vida e involuciona antes del año. Suele ser asintomático, aunque tiene tendencia a la creación de fístulas intraparenquimatosas y al desarrollo de insuficiencia cardíaca. También podemos encontrar una trombocitopenia o anemia moderada3,19,20,42,52.
- 2.
Hemangioma hepático multifocal: es el más frecuente dentro de los hemangiomas hepáticos y corresponden al HI en fase proliferativa. Es GLUT-1 positivo y suele aparecer en el contexto de una hemangiomatosis neonatal difusa o asociado con algún síndrome como el de Beckwith-Wiedemann o el de hemihipertrofia. A menudo son asintomáticos, aunque en algunos casos tiene tendencia al desarrollo de insuficiencia cardíaca e hipotiroidismo3,8,18,20,42.
- 3.
Hemangioma hepático difuso: tiene similares características que el anterior, pero afecta a la totalidad del parénquima hepático (hepatomegalia masiva), por lo que su comportamiento es mucho más agresivo, y puede provocar un síndrome compartimental abdominal, típicamente alrededor de los 3–4 meses de vida, que condiciona problemas de la ventilación, del retorno venoso y compresión de la vena renal, con fallo hepático que puede obligar a precisar un trasplante hepático. Es frecuente su asociación con un hipotiroidismo profundo, ya que estas lesiones muy a menudo expresan iodotironina deiodinasa tipo 3 que degrada la hormona tiroidea. La mayoría no presenta hemangiomas cutáneos asociados o sólo muy pocos8,42,52.
- •
Hemangiomatosis neonatal: en algunos niños se observan múltiples hemangiomas eruptivos (más de 5–10), cupuliformes, de pocos milímetros, que afectan la piel y las mucosas. Estas lesiones pueden asociarse con hemangiomas hepáticos, gastrointestinales, pulmonares o intracraneales (hemangiomatosis neonatal difusa) o pueden aparecer exclusivamente en la piel (hemangiomatosis neonatal benigna). El riesgo de afectación visceral es proporcional al número de lesiones cutáneas, sobre todo si afectan las mucosas. La tríada clínica típica de la forma difusa es hepatomegalia, insuficiencia cardíaca y anemia. El pronóstico de la forma difusa es sombrío (el 40–80% de mortalidad), ya que con frecuencia existen cortocircuitos arteriovenosos hepáticos que dan lugar a insuficiencia cardíaca grave o hemorragias internas en las primeras semanas de vida7,8,18,42,48.
- •
A diferencia de las malformaciones vasculares, los hemangiomas excepcionalmente se asocian con síndromes dismórficos53, en cambio sí se pueden asociar con diversas malformaciones:
- •
Los hemangiomas lumbosacros pueden ser marcadores de disrafias espinales (médula anclada o lipomeningocele) y alteraciones anogenitourinarias o esqueléticas subyacentes (ano imperforado, fístulas anogenitales, malformaciones renales o genitales, espina bífida, etc.). El riesgo de estas malformaciones persiste, aunque el hemangioma haya involucionado. Cuando existe una médula anclada, durante el crecimiento del niño, pueden aparecer paraplejia, pie equino y problemas esfinterianos7,8,24,39,54.
- •
La existencia de un hemangioma segmentario, sobre todo en las niñas, se puede asociar con malformaciones estructurales subyacentes (el 20% de los casos de hemangiomas segmentarios faciales), y puede afectar al sistema nervioso central, al sistema cardiovascular, al sistema musculoesquelético y al ojo25,54–57. El síndrome de PHACE(S) es el mejor ejemplo de la variedad de complicaciones que se pueden asociar con los hemangiomas segmentarios. Es un síndrome neurocutáneo poco frecuente, más frecuente en niñas y que se caracteriza por la presencia de malformaciones intracraneales de la fosa posterior (en especial, síndrome de Dandy-Walker); hemangioma facial segmentario (frontonasal, mandibular, maxilar o frontotemporal); anomalías arteriales del polígono de Willis y de los troncos supraaórticos; coartación de aorta y defectos cardíacos (tetralogía de Fallot, comunicación interventricular, etc.); anomalías oculares (Eye) (cataratas congénitas, microftalmia, coloboma, embriotoxon, hipoplasia o agenesia del nervio óptico, exoftalmos, etc.), y malformaciones de la línea media esternal (fisura o agenesia esternal, sinus dérmico torácico o ectopia cordis) o abdominal (rafe supraumbilical). Algunos pacientes también pueden tener hemangiomas subglóticos. Para su diagnóstico se necesita la existencia del hemangioma facial segmentario y una manifestación extracutánea. Más del 50% de los pacientes con anomalías cerebrales vasculares desarrollan en los primeros 5 años de vida secuelas neurológicas7,45,55,58.
Aparecen totalmente desarrollados en el momento del parto, ya que la fase proliferativa se lleva a cabo exclusivamente intraútero. Son igual de frecuentes en ambos sexos. Se manifiestan como tumores más violáceos que los HI, con telangiectasias y algunas flebectasias, tumores compactos y lobulados, nódulos duros abollonados de color rosado y con halos pálidos, o placas violáceas infiltradas con un halo azulado y lanugo6,7,34,59. Su aspecto clínico obliga a descartar otros tumores de partes blandas del recién nacido (rabdomiosarcoma embrionario, fibrosarcoma congénito, etc.)7,60,61.
La inmunorreactividad negativa frente al GLUT-1 permite distinguirlos del HI, lo que es difícil desde el punto de vista clínico e histológico en los primeros días de vida (algunos HI, especialmente en prematuros, están ya presentes en el momento del parto)7,15,17.
Los grupos de trabajo de Boston, París y Arkansas confirmaron, en 1996 y 1999, la existencia de RICH y de NICH:
- •
RICH: entidad histológica y clínica diferente, que previamente se confundía con otros tumores o malformaciones vasculares. Su involución es más o menos completa durante el primer año de vida y las secuelas postinvolutivas van desde atrofia leve a piel redundante62–64. Recientemente, se ha descrito que pueden provocar una trombocitopenia y coagulopatía transitoria65.
- •
NICH: se caracteriza por la ausencia de cambios significativos durante la infancia (crecen progresivamente con el niño sin involucionar con el tiempo). Se presentan como placas o tumoraciones poco elevadas, redondeados u ovoides, de tamaño generalmente reducido, duras al tacto, violáceas y de temperatura más elevada que la piel adyacente; lo que indica flujo sanguíneo elevado, que se demuestra por ecografía-Doppler. De hecho, inicialmente se clasificaron como malformaciones arteriovenosas superficiales17,59,66,67.
Ambos son tumores vasculares y probablemente son parte de un espectro y no una sola entidad. Hay pacientes en los que coexisten HI con HC, lo que puede ser una coincidencia (pues el HC es raro y el HI es muy frecuente) o que exista un vínculo entre éstos. A favor de esto último, hay evidencia acumulada de que las mutaciones somáticas en una única célula endotelial progenitora, con expansión clonal causan los hemangiomas, quizás diferentes mutaciones sean causantes del comportamiento divergente, con rápida involución o no involución, o la ausencia de GLUT-1 en los HC15,30,62,68. También hay quien piensa que los NICH son el estadio tardío de los RICH, aunque para otros autores son histológicamente diferentes. ¿Es posible que exista un vínculo o una asociación entre HC y HI?, ¿están relacionados ambos tipos o son dos entidades distintas? y ¿todos los HC tienen un inicio prenatal con comportamiento posnatal divergente? Hay pacientes con HC que desarrollan HI y HC que inician evolución de RICH, pero luego quedan como NICH. Mientras se unifica o se descubre el origen de éstos, se consideran entidades clínicas diferentes, con pronóstico y tratamiento distintos7,17,62,66,68.
Hemangioendotelioma kaposiformeEl término HE incluye diferentes neoplasias de partes blandas de origen vascular:
- •
HE maligno, que probablemente debe considerarse como un angiosarcoma.
- •
HE epitelioide, de malignidad intermedia, localizado en las partes blandas o en el hueso, el pulmón o el hígado.
- •
HE kaposiforme, que es una tumoración de bajo grado.
El HEK es una entidad excepcional que afecta exclusivamente a niños. Es frecuente su localización en el retroperitoneo, pero también puede afectar a la piel en forma de placas o tumores eritematovioláceos infiltrados al tacto. Se suele presentar como una lesión vascular de crecimiento rápido e intensivo en un niño menor de 3 meses, aunque algunos casos son congénitos. Mediante estudios de imagen se puede demostrar su carácter infiltrativo y la biopsia cutánea muestra hallazgos combinados de los HI y del sarcoma de Kaposi. No metastatiza y su pronóstico depende del tamaño y la localización de la lesión. Las lesiones de gran tamaño que afectan las partes blandas profundas, el mediastino o el retroperitoneo son irresecables y pueden provocar la muerte si se complican con el síndrome o fenómeno de Kasabach-Merritt, asociación muy frecuente en este tipo de tumor vascular7,69–72.
Angioblastoma o angioma en penachoSuele aparecer en el primer año de vida, aunque en un 15% de los casos está presente en el nacimiento y, en ocasiones, en la edad adulta; sin embargo, también es posible la autoinvolución, sobre todo en los congénitos. Se caracteriza por máculas, placas o tumores violáceos, únicos o múltiples, mal delimitados, infiltrados al tacto, que suelen tener un componente nodular profundo (pueden extenderse hacia el tejido celular subcutáneo, fascia y músculo). A veces es un tumor doloroso, con hiperhidrosis localizada y lanugo en su superficie. Se localiza principalmente en la zona superior del tronco/espalda, el hombro y el cuello. Presenta un crecimiento lento pero progresivo y posterior estabilidad. No suele involucionar espontáneamente. Es capaz de provocar el síndrome de Kasabach-Merritt, pero en un porcentaje mucho más bajo que el HEK7,41,73,74.
Granuloma piógenoTambién llamado hemangioma capilar lobular, granuloma telangiectásico o botriomicoma, es una lesión frecuente que afecta a niños, a adultos jóvenes y a pacientes que reciben tratamiento sistémico con derivados sintéticos de la vitamina A. Existe controversia sobre si constituye una verdadera neoplasia benigna o un proceso vascular reactivo, asociado con traumatismos o factores hormonales. Su aspecto clínico típico es el de una pápula o nódulo ligeramente pediculado, entre 0,5–2cm de diámetro, de color rojo intenso y consistencia friable, que sangra con facilidad y progresa en pocas semanas. Se localiza principalmente en las zonas expuestas a traumatismos, como los dedos de las manos, antebrazos, cara y, a veces, en mucosa oral. El estudio histológico muestra un tumor capilar lobular bien delimitado e inmerso en un estroma edematoso, con un infiltrado inflamatorio mixto. La superficie está ulcerada o recubierta por una epidermis aplanada y la base de la lesión está rodeada por un collarete epidérmico. En fases iniciales es indistinguible del tejido de granulación y en estadios evolucionados corresponde a tejido cicatricial75–77.
Diagnóstico de los tumores vasculares infantilesAproximadamente un 90% de los hemangiomas se diagnostican fácilmente en función de las características clínicas previamente expuestas, no tienen complicaciones y no se asocian con alteraciones extracutáneas, por lo que no es preciso solicitar exploraciones complementarias. De hecho, en toda anomalía/lesión vascular cuyo diagnóstico no pueda establecerse con seguridad en los primeros meses de vida y que no plantee necesidad de usar tratamiento se aconseja una actitud expectante3,7,11,23,38,78.
Sin embargo, pueden realizarse exploraciones complementarias no invasivas en el período de observación en los siguientes casos8:
- •
Si existen dudas diagnósticas de estar ante un HI.
- •
Para valorar la extensión cutánea y extracutánea del tumor.
- •
Para seguir la evolución espontánea o la respuesta al tratamiento.
- •
En caso de complicaciones, tales como la hemorragia de hemangiomas viscerales.
- •
Cuando puedan existir malformaciones asociadas.
Los estudios habitualmente más recomendados y utilizados son los siguientes:
1. Estudios de imagen:
1.1. Ecografía simple combinada con imágenes Doppler en color y análisis espectral Doppler7,42,52,54,61,67: sirven para valorar el contenido tisular y las características del flujo de las lesiones vasculares. Son técnicas no invasivas, relativamente baratas, pero que no permiten valorar con precisión la extensión de las lesiones y dependen de la experiencia del ecografista. Las indicaciones más aceptadas son las siguientes:
- •
Establecer el diagnóstico diferencial con otros tumores de partes blandas y malformaciones vasculares de bajo flujo.
- •
En niños menores de 4 meses con hemangiomas lumbosacros, con el fin de valorar disrafismo oculto y malformaciones genitourinarias.
- •
Descartar hemangiomas viscerales en la hemangiomatosis neonatal. La ecografía hepática está indicada cuando existen 5 o más hemangiomas cutáneos y la ecografía cerebral en lactantes pequeños con hemangiomas segmentarios faciales.
- •
Seguimiento evolutivo de tumores vasculares en tratamiento o de los hemangiomas hepáticos focales y multifocales asintomáticos para valorar su involución.
1.2. Resonancia magnética (RM) sin o con gadolinio±angiorresonancia7,8,11,25,36,41,42,44,52,58,63,79–81: es la técnica de elección para el estudio de las lesiones vasculares cutáneas. Permite valorar la naturaleza y la extensión exacta de éstas, lo que es muy importante previo a tomar decisiones terapéuticas. Indicaciones:
- •
Hemangiomas lumbosacros, si la ecografia es patológica o en mayores de 4 meses para descartar disrafismo oculto o extensión intraespinal.
- •
Hemangiomas perineales extensos, ya que se pueden asociar con anomalías urogenitales o anales.
- •
Hemangiomas segmentarios de la cabeza y del cuello que pueden asociar malformaciones estructurales y vasculares cerebrales, así como daño de la vía aérea.
- •
Hemangiomas hepáticos sintomáticos.
- •
Hemangiomas de presentación atípica (ante la sospecha de hemangioma, pero en presencia de una trombocitopenia grave debe descartarse un HEK).
- •
Lesiones de alto flujo, con dudas en la ecografía sobre si es un RICH o una malformación arteriovenosa.
2. Pruebas de laboratorio:
El hemograma y el estudio de coagulación pueden ser útiles para valorar anemia aguda o crónica por hemorragia de las lesiones cutáneas o viscerales (sobre todo digestivas en la hemangiomatosis neonatal difusa) o trombocitopenia moderada en hemangiomas hepáticos localizados o trombocitopenia grave asociada con hipofibrinogenemia y aumento del dímero D en el síndrome de Kasabach-Merritt2,3,19,51,72,82.
Está indicado realizar estudio de hormonas tiroideas (T4 libre y TSH) en hemangiomas extensos, sobre todo en los hepáticos multifocales y difusos8,42.
La determinación de niveles urinarios altos del bFGF es un marcador de angiogénesis activa. Su determinación mediante ELISA en muestras de orina aislada, expresada en picogramos de bFGF por gramos de creatinina, puede ser útil para diferenciar los hemangiomas en fase proliferativa de malformaciones vasculares, valorar si el hemangioma o el HEK están en fase proliferativa y, sobre todo, para monitorizar la respuesta al tratamiento médico con interferón (INF)83.
3. Endoscopia:
La laringoscopia puede estar indicada para valorar si existe obstrucción de la vía aérea en hemangiomas de la vía aérea7,8,45,47. La endoscopia digestiva es útil en pacientes con hemangiomatosis neonatal difusa con hemangiomas sangrantes en el tracto digestivo7,51.
4. Estudio oftalmológico:
Este estudio es recomendable en los hemangiomas palpebrales, ya que pueden ocluir la hendidura palpebral, distorsionar la córnea, y dañar la visión del niño. También debe indicarse un examen oftalmológico cuando se trata de hemangiomas cervicofaciales de gran tamaño3,7,8,46.
5. Estudio cardiológico:
En hemangiomas cervicofaciales o torácicos de gran tamaño para descartar asociación con coartación de aorta u otras malformaciones vasculares, y en hemangiomas hepáticos que presenten insuficiencia cardíaca7,48,55.
6. Biopsia/estudio histológico:
Es la prueba diagnóstica definitiva. Se debe considerar realizarla en los siguientes casos:
- •
Ante la presencia de una lesión vascular cuyo diagnóstico clínico no pueda establecerse con seguridad en los primeros meses de vida y que plantee la necesidad de utilizar medidas terapéuticas activas a muy corto plazo. Histológicamente, los hemangiomas presentan una hiperplasia vascular y una proliferación de las células endoteliales, y es muy aconsejable realizar el marcador inmunohistoquímico GLUT-1, positivo en el HI y negativo en el HC. Este patrón de positividad se mantiene en todas las fases del HI, incluida la involutiva7,14–17,38,66.
- •
El angioblastoma y el HEK tienen un patrón histológico característico además de ser GLUT-1 negativos70,71,73.
- •
Nos permite realizar el diagnóstico diferencial con tumores de aspecto vascular que en realidad son otros tipos histológicos60,77,84.
Los HI y los HC deben distinguirse principalmente de las malformaciones vasculares (fig. 1). Estas últimas son lesiones benignas, no tumorales (alteraciones estructurales que originan un defecto de la constitución de la pared del vaso —vasos displásicos—), presentes siempre desde el nacimiento; aunque a veces no son visibles hasta semanas o meses después, con un crecimiento proporcional al corporal del niño (no involucionan), con igual frecuencia en ambos sexos, a menudo asociadas con síndromes dismórficos y GLUT-1 negativas2–4,7,11–14,35,36,85,86. En ocasiones, nos podemos plantear el diagnóstico diferencial con otros tumores vasculares infantiles como el HEK y el angioblastoma, u otros de aspecto vascular como el fibrosarcoma congénito, rabdomiosarcoma, miofibromatosis-hemangiopericitoma, teratoma, glioma nasal, lipoblastoma, dermatofibrosarcoma protuberans o neurofibroma3,7,60,70,73,84.
Si sospechamos un hemangioma hepático, pero presenta un patrón radiológico anómalo o no concluyente, deberemos siempre descartar que no se trate de un hepatoblastoma, hamartoma mesenquimal o metástasis de un neuroblastoma52.
Tratamiento de los tumores vasculares infantilesLa mayoría de los hemangiomas (80–90%) no va a requerir tratamiento3,7,8,87. Si consideramos el impacto psicológico que una tumoración vascular visible provoca en el desarrollo del niño y en sus padres durante un período prolongado, y la mínima morbilidad que en la actualidad representan algunos tratamientos (los esteroides o la extirpación quirúrgica), la postura de abstención terapéutica puede ser la más cómoda, pero no la más indicada de forma generalizada41,88–90.
Objetivos8- •
Prevenir o tratar complicaciones que pongan en peligro la vida o la función de un órgano.
- •
Prevenir la desfiguración permanente.
- •
Minimizar el estrés psicosocial al niño y a su familia.
- •
Evitar procedimientos intensivos, con secuelas potenciales.
- •
Prevenir o tratar la ulceración para minimizar la infección, dolor o cicatrización.
- 1.
Actitud expectante: indicada en niños asintomáticos (hemangiomas pequeños, localizados lejos de zonas con posible daño funcional y velocidad de crecimiento lenta, y en el RICH)7,59,62,87,89.
Además de evaluar al paciente, se debe tener en cuenta la opinión de los padres. En el seguimiento por consulta se recomienda en la primera visita explicar la evolución natural de la lesión y su pronóstico, discutir las ventajas y las desventajas de los distintos tratamientos, revisiones clínicas frecuentes (cada 15–30 días) en los niños más pequeños o cada 1–2 meses hasta que los hemangiomas inicien su regresión, medir y fotografiar la lesión para constatar su ritmo de crecimiento y eventual involución, mostrar la evolución de casos semejantes y apoyo emocional8.
- 2.
Tratamiento activo: indicado en niños sintomáticos (cualquier caso que implique amenaza para la vida, para la función de órganos próximos o que pueda provocar secuelas importantes)3,7,8,24,41–43,45,46,48,59,67,72,87,89–96, como en el caso de:
- •
Hemangioma periorbitario que provoca trastorno de la función visual (oclusión del eje visual por un tumor palpebral, compresión del nervio óptico por un rápido crecimiento, exposición corneal excesiva debida a una proptosis grave o anisometropía inducida por el tumor).
- •
Hemangioma subglótico de tamaño medio con reducción de la vía aérea inferior al 50%, sin extensión extralaríngea constatada por RM.
- •
Hemangioma del canal auditivo externo que puede producir alteración de la función auditiva y como consecuencia un retraso en la adquisición del lenguaje.
- •
Hemangioma de la región anogenital con alteración de la función intestinal o urinaria.
- •
Complicaciones como hemorragia, ulceración (zona del pañal, zona superior de la espalda, cuello, cartílago nasal o auricular, labios o extremidades), infección secundaria o dolor.
- •
Hemangiomas de crecimiento rápido que produzcan o puedan producir deformidad de la cara (párpado, canal auditivo, nariz o labios) o de otras localizaciones.
- •
Afectación importante de la estética.
- •
Lesiones con afectación visceral que provocan una ICC (hemangioma hepático) o una hemorragia grave (hemangioma gastrointestinal).
- •
HEK y angioblastoma asociados o no con el síndrome de Kasabach-Merritt.
- •
Problemas psicológicos en los familiares o en el paciente, debido a la presencia del tumor o sus secuelas.
- •
Dentro del tratamiento activo hay varias posibilidades e indicaciones para cada una de éstas:
2.1. Tratamiento quirúrgico: antes de marcar la indicación quirúrgica (discutida y consensuada entre el equipo multidisciplinario), hay que tener en cuenta 3 puntos importantes: debe ser bajo consenso entre la familia y el equipo médico, hay que valorar los criterios de resecabilidad y la toma de decisión debe ser precoz.
Las indicaciones más reconocidas son hemangioma periorbitario, parotídeo, labial, punta de la nariz, NICH, angioblastoma, HEK (si las condiciones clínicas del paciente lo permiten) y para la corrección de las deformidades estéticas residuales7,8,24,41,43,46,59,66,73.
Una vez que la indicación quirúrgica se establece, hoy en día no hay ninguna explicación con base científica que justifique retrasar una intervención necesaria, pues se conoce la mayor capacidad de cicatrización del lactante respecto al niño mayor o al adulto. Si se indica en la fase involutiva es importante que se haga lo más precoz posible, y para así evitar secuelas psicológicas a largo plazo, ya que la formación de la imagen facial ocurre a la edad de 2,5–3 años8,90.
2.2. Tratamiento farmacológico: indicado en el resto de las situaciones previamente incluidas en las indicaciones generales de tratamiento activo, teniendo en cuenta las diferentes opciones existentes según cada caso y evolución (fig. 3):
- •
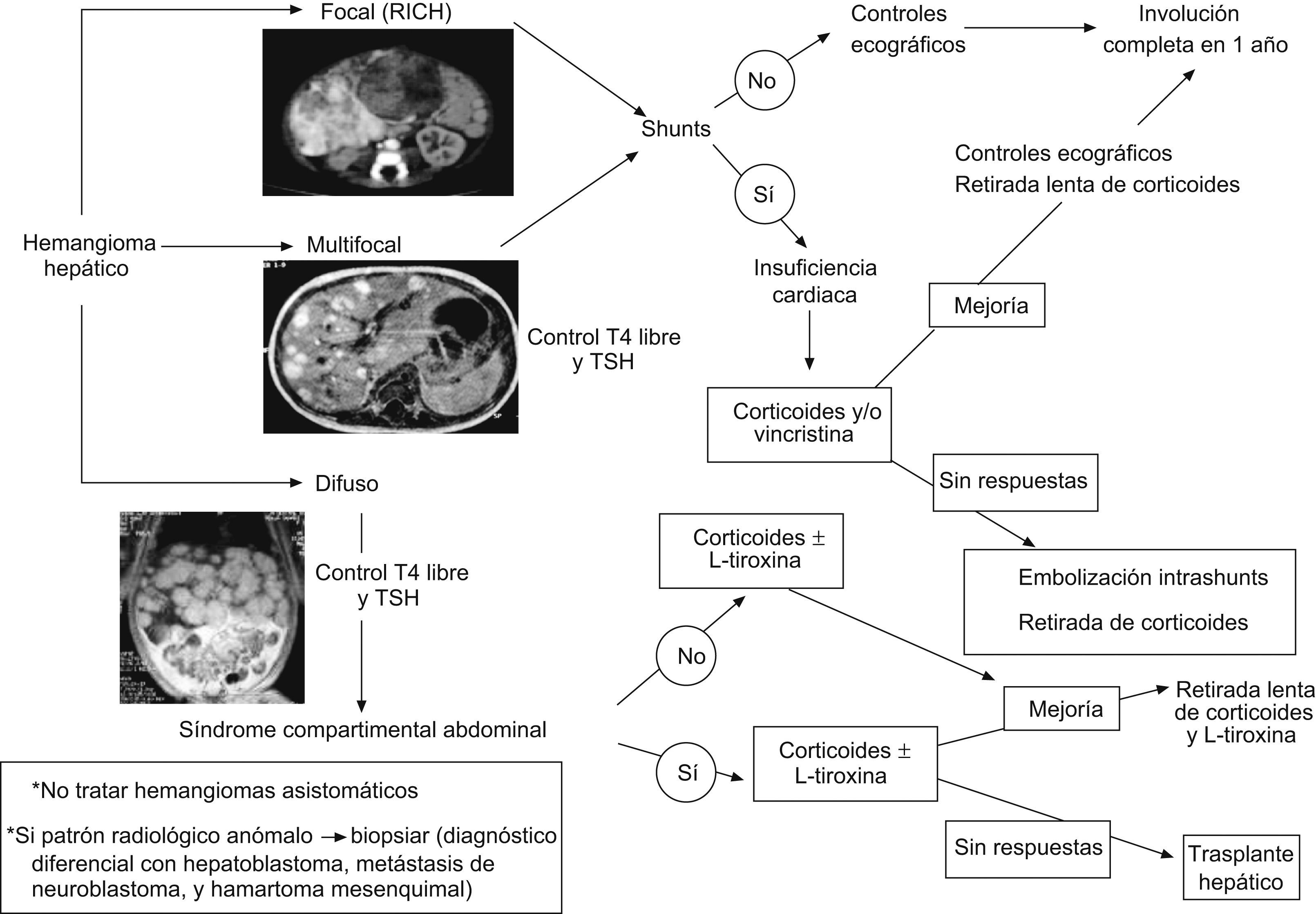
Corticoides sistémicos: debe utilizarse prednisona o prednisolona oral en una dosis no inferior a 2–3mg/kg/día (5mg/kg/día en el caso de hemangiomas graves como aquellos que obstruyen la vía respiratoria y en el síndrome de Kasabach-Merritt), en dosis única diaria administrada por la mañana. Es importante comenzar el tratamiento siempre antes de los 6 meses de vida, para aprovechar una mejor respuesta en la fase proliferativa2,3,7,8,24,71,89,92,93,96. En el caso de los hemangiomas subglóticos pueden requerir intubación-ventilación mecánica para mantener permeable la vía aérea hasta que se inicia el efecto8,45,90. Los lactantes los suelen tolerar bien, aunque hay que tener en cuenta sus efectos secundarios más frecuentes en estos casos (inmunosupresión transitoria, molestias gastrointestinales, alteraciones del sueño y aumento del apetito), que son generalmente temporales96,97. Hay que revaluar su respuesta a las 2–3 semanas8,42,49,50,92,94–96:
- ○
Si se produce respuesta positiva, al comprobar que la lesión se ablanda, palidece y cesa su crecimiento (mejoría en un tercio de los casos o estabilización del crecimiento en otro tercio, durante los primeros 7–10 días de iniciado el tratamiento con corticoides): mantener la dosis inicial durante 4 semanas más y después disminuir lentamente hasta suspenderlos definitivamente 10 semanas más tarde.
- ○
Si durante la retirada de los corticoides se produce un recrecimiento del hemangioma (referido hasta en un 36% en distintas series): reinstaurar la dosis previa eficaz y mantenerla durante 4 semanas, reevaluar entonces la respuesta, y si fue favorable, se vuelven a retirar progresivamente.
- ○
Si no existe respuesta inicial (hasta en un tercio del total de los casos, en ocasiones debida a una dosis insuficiente de corticoides): suspender el tratamiento con corticoides y en función del daño clínico y la edad del paciente comenzar con INF o vincristina.
- ○
- •
Corticoides intralesionales: es un tratamiento efectivo en indicaciones muy seleccionadas, que consigue evitar los efectos adversos de los corticoides sistémicos y su acción es más rápida (24–48h). Sus posibles indicaciones son las siguientes7,45,46,89,91,94,98:
- ○
Hemangioma cutáneo pequeño bien delimitado a nivel facial con localización problemática (párpado).
- ○
Hemangioma cutáneo que progresa a pesar de haber respondido inicialmente a los corticoides sistémicos.
- ○
Hemangioma subglótico sin respuesta al resto de los tratamientos médicos o cuando éstos son imposibles de administrar.
- ○
Se recomienda administrarlos bajo sedoanalgesia, para así evitar el movimiento del paciente durante su inyección. Se suele utilizar el acetato de triamcinolona y el acetato de betametasona o dexametasona, en jeringas separadas de 2ml, que se administrarán intralesionalmente en diferentes direcciones a través de una punción única o varias punciones, según el tamaño de la lesión y sin presión. Se pueden repetir las inyecciones con un intervalo medio de 4–8 semanas, hasta un máximo de 7 inyecciones. La respuesta clínica se observa en los primeros 3 días (blanqueamiento y posterior regresión de la masa). Marcada regresión en el 64%, moderada en el 24% y mínima en el 1%. La respuesta guarda relación con el volumen del hemangioma (si es superior a 20cm3, presentará una peor respuesta)98.
En el caso de los hemangiomas subglóticos, precisarán intubación nasotraqueal inmediata tras su administración y el paciente suele mantenerse libre de síntomas obstructivos durante 9 meses. Si recurre la clínica de obstrucción, puede repetirse el tratamiento hasta 3 veces. La ventaja respecto a la cirugía es la ausencia de desarrollo de estenosis subglótica91.
- •
INF α 2a o 2b subcutáneo: es un tratamiento igual de efectivo a cualquier edad y no sólo en la fase proliferativa. Su efectividad es mayor cuanto antes se inicie. La indicación más extendida para su uso ha sido en lesiones con riesgo vital o funcional grave, que no han respondido previamente a corticoides, teniendo en cuenta que está contraindicado en menores de 6 meses, y a valorar individualmente entre 6 y 12 meses, ya que en estas edades el riesgo de diplejía espástica relacionada con este tratamiento es más elevado. El 60% suele responder (más en hemangiomas de mejilla o parótida) con una reducción del tamaño de la lesión del 75%3,7,8,24,40,49,50,72,89,95,99–102.
Se suele comenzar con una dosis de 1 millón de UI/m2/día durante 3 días, y se continúa con 2 millones de UI/m2/día durante otros 3 días, para posteriormente pasar a 3 millones de UI/m2/día, hasta obtener la respuesta deseada. La respuesta al INF se evalúa tras 3 meses de tratamiento en dosis completa8:
- ○
Si hay buena respuesta (masa inferior a un tercio del tamaño inicial y <5cm) se suspende el tratamiento.
- ○
Si hay respuesta parcial (masa superior a un tercio del tamaño inicial y <5cm) se mantiene el INF hasta completar 6–10 meses.
- ○
Si hay ausencia de respuesta se suspende y se comienza con vincristina.
- ○
- •
Vincristina: presenta una tasa de respuesta cercana al 100% en HI. No se ha constatado rebote tras suspenderla. Se considera como una segunda opción terapeútica en la mayoría de los centros a nivel nacional y mundial. Sus indicaciones más reconocidas son las siguientes7,8,18,24,72,89,103–106:
- ○
Cuando el tratamiento corticoideo ha fracasado y no hay un riesgo vital o funcional inmediato en pacientes de edad ≤6–12 meses.
- ○
Hemangiomas hepáticos con insuficiencia cardíaca y si no hay respuesta a corticoides, sola o asociada con éstos.
- ○
Cuando no pueden administrarse corticoides debido a efectos secundarios graves o a imposibilidad para retirarlos completamente.
- ○
Cuando el tratamiento con INF fracasa o si está contraindicado.
- ○
- •
Ciclofosfamida: de igual forma que la vincristina, en dosis bajas actúa como inhibidor de la angiogénesis en tumores de modelos murinos. Se ha descrito efectiva en 10 casos en la literatura médica, en hemangiomas diseminados con riesgo vital no respondedores a los tratamientos previos. La pauta utilizada fue con dosis de 10mg/kg/día, 4 días por semana, cada 14 días, de 1–4 ciclos. No se han detectado efectos secundarios destacables7,89,99.
- •
Propranolol: desde hace un año aproximadamente y después de la descripción de su efecto antiproliferativo, el uso de propranolol en solución (1mg/kg/12h) se está extendiendo rápidamente a nivel experimental y todos los indicios hacen pensar que sustituirá definitivamente a los corticoides como primera opción farmacológica, aunque su indicación aún no está oficialmente aprobada. Esto se debe fundamentalmente a la menor incidencia de efectos secundarios y a un efecto más rápido que los esteroides. No obstante, como efectos secundarios puede producir hipoglucemia, hipotensión y crisis de broncoespasmo en pacientes con antecedentes de asma. Los mecanismos de acción propuestos a través de éstos ejerce su acción en los hemangiomas e incluyen el control del estrés hipóxico, inducción de la apoptosis y disminución de la producción de factor de crecimiento del endotelio vascular y del bFGF. Es efectivo tanto en la fase proliferativa como en la involutiva de los hemangiomas, y su resultado es más evidente en los hemangiomas segmentarios que en los focales107–109, y también en los subglóticos110. No se ha observado respuesta al propranolol en el angioblastoma y el HEK. En el síndrome de PHACE(S) podría aumentar el riesgo de ictus intracraneal, aunque no hay ninguna publicación al respecto hasta el momento109. Hoy en día, hay pocas dudas sobre su eficacia y ya están en marcha en países de nuestro entorno ensayos clínicos con su uso en dosis más altas.
2.3. Tratamiento fotodinámico (láser de colorante pulsado): en los últimos años el láser ha ido adquiriendo una relevancia progresiva en el tratamiento de los hemangiomas, tanto en la fase aguda como de las secuelas. De hecho, las frecuentes ulceraciones de los hemangiomas periorales o en el área genital responden de forma más rápida (<48h) y efectiva (reducción del dolor) al láser que a los esteroides7,8,40,89. Igualmente, es el tratamiento de elección en la vascularización residual postinvolutiva7,8. Queda por determinar su verdadera utilidad en la fase proliferativa de los hemangiomas superficiales, en los que su uso indiscriminado ha producido una tasa inaceptable de fracasos y de complicaciones111.
No se recomienda su uso con fines cosméticos en pacientes menores de 6 meses de edad, por el alto riesgo de ulceración8,86,112.
2.4. Embolización: su indicación más frecuente es su uso en los hemangiomas hepáticos que presentan shunts y clínica de ICC tras no responder a corticoides o vincristina. En ocasiones, puede ser un tratamiento de segunda línea en hemangiomas alarmantes, no cutáneos, tras los corticoides sistémicos y el INF o vincristina, de forma aislada o asociada con tratamiento médico o quirúrgico. Su eficacia real en hemangiomas superficiales extensos aún está por dilucidar, por esto, de momento, en estos casos no está indicada19,42,52,112.
2.5. Tratamientos tópicos experimentales: se han utilizado el imiquimod al 5% y el factor de crecimiento derivado de las plaquetas en ulceraciones, pero su uso no está aprobado en la edad infantil8.
Tratamiento en situaciones especiales- •
Tratamiento de los hemangiomas hepáticos (fig. 2): una vez diagnosticados, su evolución clínica puede predecirse por la clínica que provocan, que habitualmente aparece en las primeras semanas de vida (asintomáticos o hepatomegalia, anemia e ICC) y por los hallazgos en las técnicas de imagen (solitarios o multifocales, con/sin shunts de alto flujo y difusos)8,18,19,42,52,112.
- 1.
Hemangioma hepático solitario: el tratamiento se hace según la sintomatología:
- ○
Asintomático: observación con controles ecográficos hasta verificar involución completa.
- ○
Insuficiencia cardíaca: corticoides sistémicos o vincristina y, si no responden, pasar a embolización de shunts de alto flujo.
- ○
- 2.
Hemangioma hepático multifocal: el tratamiento también se hace según la sintomatología, como en el caso del hemangioma hepático solitario.
- 3.
Hemangioma hepático difuso: requieren ingreso en una unidad de cuidados intensivos pediátricos, por la alta mortalidad que presentan en las fases iniciales de la enfermedad. Precisan corticoides sistémicos en altas dosis (5mg/kg/día) y si existe hipotiroidismo asociado, tratamiento sustitutivo con altas dosis de L-tiroxina (oral: 15μg/kg/día, cada 24h o i.v.: el 75% de la dosis oral). Si no responden al tratamiento médico, está indicado realizar un trasplante hepático.
- 1.
- •
Tratamiento de los hemangiomas subglóticos: los localizados requieren resección quirúrgica o corticoides intralesionales. En los segmentarios con afectación bilateral/circunferencial se recomienda iniciar tratamiento con corticoides sistémicos, y si no hay respuesta, utilizar vincristina. No se recomienda la traqueostomía ni la cirugía por el alto riesgo de morbimortalidad que presentan7,8,45,47,89–91,98.
- •
Tratamiento de los HC:
- ○
RICH: actitud espectante (esperar durante unos meses la involución natural de la tumoración). En ocasiones, es necesario corregir las deformidades estéticas postinvolutivas, ya sean cicatrices o atrofias que producen asimetría facial24,59,62,63.
- ○
NICH: extirpación quirúrgica. La evolución posquirúrgica es favorable, incluso cuando la resección es incompleta, no se observan recidivas en los márgenes de resección, y es habitualmente innecesaria la embolización preoperatoria, ya que a pesar del moderado flujo vascular que presenta a su través, la hemorragia intraoperatoria es fácilmente controlable7,59,66.
- ○
- •
Tratamiento del granuloma piógeno: extirpación quirúrgica o rebanado de la lesión seguido de cauterización de la base7,75,77.
Se produce exclusivamente en el HEK y el angioblastoma cuando son mayores de 10cm, pero nunca en HI ni en ninguna otra malformación vascular7,69–71,74,82.
Es una complicación caracterizada por una coagulopatía de consumo secundaria al atrapamiento local de plaquetas en el tumor. Ocasionalmente se presenta al nacimiento, afecta sobre todo a niños menores de 3 meses y se acompaña de una elevada mortalidad (20–30%). Su presentación coincide con un aumento rápido del tamaño del tumor y presenta equimosis, trombocitopenia grave (<20×109/l), consumo de factores de la coagulación (fibrinógeno bajo, aumento del dímero D±prolongación del tiempo de protrombina y el tiempo de cefalina), y palidez cutaneomucosa (anemia hemolítica no inmunitaria)7,82,99,106. No debe confundirse con la coagulopatía asociada con algunas malformaciones venosas o mixtas7.
Este proceso requiere un tratamiento intensivo multidisciplinario, ingresado inicialmente en una unidad de cuidados intensivos pediátricos:
- •
La escisión quirúrgica del tumor es el tratamiento más efectivo, pero en raras ocasiones es posible en el HEK, aunque sí en el angioblastoma71,72,74.
- •
Corticoides sistémicos en altas dosis (5mg/kg/día) como primera opción, ver respuesta como máximo en 2 semanas, aunque raramente son efectivos solos. Si no hay respuesta al tratamiento con corticoides, añadir INF (tasa de respuesta estimada del 50–60% que se puede predecir al medir en orina el bFGF, ya que a mayor elevación de éste hay una mejor respuesta). Si no hay respuesta a lo previo, usar vincristina7,72,83,99,103,106.
- •
Como tratamiento de soporte, la ticlopidina junto con el ácido acetilsalicílico por vía oral (10mg/kg/día, cada uno) asociados con los tratamientos previos o solos, se han mostrado extremadamente eficaces para el tratamiento de la trombocitopenia. El fibrinógeno o el plasma fresco congelado pueden ayudar a controlar la coagulopatía, pero no así las transfusiones de plaquetas, ya que se consumen con extremada rapidez y pueden provocar aumento del tamaño del tumor y empeorar la situación clínica del niño. Sólo transfundir plaquetas si hay hemorragias activas o antes de los procedimientos quirúrgicos o las canalizaciones venosas centrales7,72,106.
- •
De forma esporádica se han descrito respuestas a la ciclofosfamida, a la pentoxifilina, al ácido ε-aminocaproico, al ácido tranexámico y a la radioterapia. La embolización arterial sólo es útil como coadyuvante preoperatorio y en los tumores inoperables desarrolla arterias colaterales sin conseguir a largo plazo mejora de la sintomatología7,99.
Las lesiones residuales tras la resolución de la coagulopatía son relativamente frecuentes69–72.
Conclusiones- •
No debe denominarse “angioma” a cualquier lesión vascular.
- •
El dato de más valor en la historia clínica es la forma de aparición y evolución de la lesión vascular en los primeros 3–6 meses de vida.
- •
Ante una duda diagnóstica, la ecografía-Doppler, el hemograma y coagulación, la RM y la biopsia son los pasos sucesivos para confirmar el tipo de lesión vascular antes de tomar una decisión terapéutica.
- •
La mayor parte de los tumores vasculares, sobre todo los HI, no precisan tratamiento. En los que esté indicado, éste debe individualizarse. Hoy en día disponemos de diferentes tipos de tratamiento de reconocida eficacia (quirúrgico, farmacológico con distintos agentes antiangiogénicos como corticoides, INF o vincristina, o el novedoso propranolol, fotodinámico y endovascular), y la elección de uno u otro debe hacerse en función de una indicación concreta y protocolizada.
- •
Correctamente diagnosticados y en el contexto de un equipo multidisciplinario, se pueden obtener unos resultados terapéuticos altamente efectivos.
Los autores declaran no tener ningún conflicto de intereses.
