


El xantogranuloma juvenil (XJ) es una entidad descrita por primera vez hace más de un siglo por Adamson y es la forma más común de histiocitosis de células no Langerhans. Normalmente, se presenta durante la infancia (al nacimiento o dentro de los dos primeros años de vida). Es de carácter benigno y generalmente autorresolutiva en meses a años.
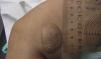
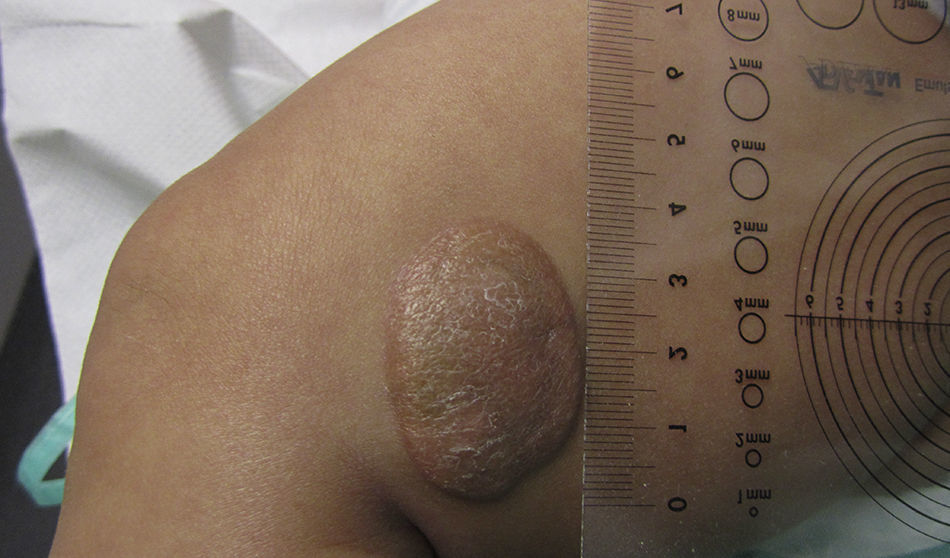
Presentamos el caso de un paciente varón de 2 meses de vida, sin antecedentes familiares ni perinatales de interés, que consulta por presentar una lesión en la pierna izquierda de 15 días de evolución, de superficie lisa brillante y color rojizo de 30 x 23mm de diámetro. Es derivada al servicio de dermatología, donde se realiza biopsia. El estudio anatomopatológico es informado como xantogranuloma de tipo mononuclear con inmunorreactividad de las células frente a CD68, CD 163, MAC 387 y CD 16 y negatividad frente a S100, CD1a y CD34. Se realizan una ecografía abdominal y un examen oftalmológico, que descartan afectación sistémica, y no han aparecido otras enfermedades asociadas. Debido a las dimensiones de la lesión y ante las posibles secuelas estéticas de la extirpación, se decide conducta expectante a la espera de su involución espontánea. Trascurridos 3 meses del diagnóstico la lesión mide 39 x 26mm (fig. 1), adquiriendo una coloración pardo-anaranjada típica. Actualmente, está siendo controlada en las consultas de dermatología y pediatría.
El XJ es una histiocitosis de células no Langherhans, benigna, de patogenia desconocida. Se caracteriza por pápulas o nódulos de 1-20mm, normalmente solitarios (60-80% de los casos)1, de consistencia firme; las lesiones son inicialmente rosa-rojas y posteriormente adquieren la coloración típica pardo-anaranjada. Se localizan principalmente en cara, cuello, tronco superior y, con menos, frecuencia en raíz de las extremidades2.
La incidencia real del XJ es desconocida; ocurre predominantemente en la infancia (hasta el 17% son congénitos y hasta un 70% se presentan antes del primer año de vida)3. No hay diferencias en la distribución por sexos, aunque en los casos de XJ múltiple la predisposición por el sexo masculino es de 5:14. No se ha descrito asociación familiar y es más frecuente en la raza blanca que en la negra.
Los XJ pueden clasificarse en micronodulares (de 2-5mm) y macronodulares (5mm a 2cm.). El XJ gigante2 es una variante poco frecuente, que mide de 2 a 10cm, de aparición congénita o en los primeros meses de vida.
El diagnóstico es clínico, aunque en ocasiones requiere confirmación histológica mediante biopsia. El patrón dermatoscópico5,6 en «sol poniente» revela un anillo periférico oscuro, con una zona central naranja-amarilla y vasos puntiformes en esta.
La histología de las lesiones presenta una acumulación de histiocitos entremezclados con células gigantes de Touton. El estudio inmunohistoquímico permite establecer el diagnóstico diferencial con la histiocitosis de células de Langerhans. El XJ expresa CD68, factor XIIIa, y es negativo para CD1a y S100, marcadores característicos de la de Langerhans2,6.
El diagnóstico diferencial4 ha de establecerse con: urticaria pigmentosa, histiocitosis cefálica, xantomas tuberosos, histiocitoma, reticulohistiocitosis congénita, dermatofibroma, linfocitoma, mastocitosis, angiomas y nevus.
El estado de salud del niño no está afectado y su desarrollo físico y psicomotor es normal, salvo si hay complicaciones por su localización visceral o por asociación con otras enfermedades4.
Los hallazgos extracutáneos7,8 son poco frecuentes, siendo el ojo el órgano más afectado (0,4%), pudiendo producir efecto masa, glaucoma, hifema e incluso ceguera. Dicha afectación ocular ocurre principalmente en pacientes menores de 2 años con lesiones múltiples (92%)8; en estos casos, podría estar indicada la revisión oftalmológica cada 6 meses hasta los 2 años de edad.
También se han descrito casos excepcionales con lesiones en pulmón, hígado, bazo, sistema nervioso central y músculo8, así como una posible asociación con leucemia mielomonocítica juvenil en pacientes con neurofibromatosis 1 que desarrollan xantogranulomas8. Sin embargo, el cribado de una posible patología visceral o hematológica asociada debe ir guiado por la clínica y no realizarse indiscriminadamente en todos los pacientes con una forma aislada de XJ8,9,10.
Las lesiones cutáneas pueden dejar como secuelas atrofia o lesiones semejantes a anectodermia8.
El tratamiento7,8 es conservador, adoptando una actitud expectante, dado que el curso es benigno y con tendencia a la regresión espontánea en meses-años, sobre todo en las formas múltiples o en las gigantes, como en nuestro caso, cuyas lesiones pueden alcanzar los 10cm de diámetro, evitando así las secuelas estéticas de la intervención.
La biopsia es necesaria en caso de duda diagnóstica, como en nuestro caso, siendo en las formas micro y macronodulares (menores de 2cm), además, terapeútica7.
En los casos excepcionales de afectación sistémica, está indicada la poliquimioterapia8.
