

Acute intermittent porphyria (AIP; MIM# 176000) is an autosomal dominant genetic disease with incomplete penetrance (10–15%) and variable expression. It is caused by a partial deficiency of the hydroxymethylbilane synthase (HMBS) enzyme resulting from mutations in the HMBS gene. Recently, a prevalence of 6.3 cases per million inhabitants has been reported for Spain,1 but a higher prevalence is estimated for the Region of Murcia due to the founder effect of the c.669_698del mutation.2
It typically manifests as intermittent attacks of abdominal pain accompanied by other gastrointestinal, cardiovascular, neurological and/or psychiatric symptoms, and many precipitating factors are known, such as prolonged fasting or the consumption of alcohol or certain drugs (Table 1).
Precipitating drugs.
| Aminoglutethimide | Barbiturates |
| Carbamazepine | Chloramphenicol |
| Clemastine | Clonidine |
| Co-trimoxazole | Danazol |
| Dapsone | Dihydralazine |
| Dimenhydrinate | Metamizole |
| Ergot derivatives | Erythromycin |
| Etamsylate | Ethosuximide |
| Etomidate | Griseofulvin |
| Ketoconazole | Meprobamate |
| Methyldopa | Sulphonamides |
| Methysergide | Nalidixic acid |
| Orphenadrine | Oxcarbazepine |
| Tolbutamide | Phenylbutazone |
| Phenytoin | Pyrimidone |
| Pyrazolone | Pyrazinamide |
The prevalence of AIP in the paediatric population is unknown, symptomatic cases are exceptional, and the reported clinical manifestations are variable. Only one prospective study has been conducted, in Sweden, by Hultdin et al.3; it included 61 patients aged less than 18 years, of whom 10% experienced attacks (all experienced mild abdominal pain).
Although diagnosing AIP in children poses great challenges, this disease should be suspected in patients with nonspecific gastrointestinal, neurological and/or psychiatric manifestations, and should be included in the differential diagnosis of abdominal pain of unknown origin. When suspected, the preferred screening method is the Hoesch test (qualitative detection of PBG in urine), increasingly used in Spain as a result of the launching of the PAGORA project (Acute Porphyria Screening Protocol for patients with unexplained acute abdominal pain in the emergency department), to be followed by specific enzymatic or genetic testing. The management is based on removal of precipitating factors, symptomatic treatment and increased intake of carbohydrates in mild to moderate cases; there is evidence supporting the efficacy of the administration of hemin as haem arginate in severe cases,4 and recently Andreeva et al. have published a note about a research project on the use of genetic therapy in these patients (2014).5
We performed a retrospective review of the medical records of patients aged less than 18 years with a molecular diagnosis of AIP after the positive identification of a family history of AIP (affected parent) during the followup by the medical genetics department. We analysed the prevalence, phenotype and genotype of these cases.
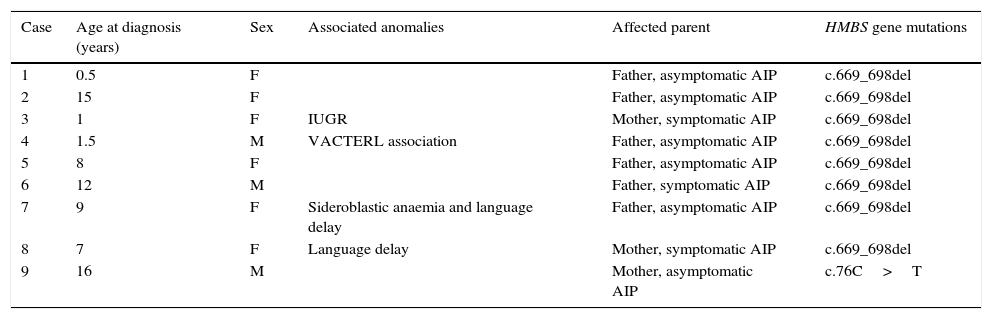
We present the cases of nine patients, with a mean age at diagnosis of 7 years (Table 2). The prevalence is 28 cases per million inhabitants (total population under 18 years in Murcia, 320,698). The mean duration of followup after diagnosis was 23 months (range, 3–40 months), and according to the documentation, none of the patients experienced attacks during the followup. At age 2, case 9 experienced an episode of mild abdominal pain after the molecular diagnosis of AIP, so he was admitted to the hospital and diagnosed with pain of unknown origin (urine PBG was not measured to assess for a possible attack).
Summary of epidemiologic characteristics, phenotype and genotype of the cases.
| Case | Age at diagnosis (years) | Sex | Associated anomalies | Affected parent | HMBS gene mutations |
|---|---|---|---|---|---|
| 1 | 0.5 | F | Father, asymptomatic AIP | c.669_698del | |
| 2 | 15 | F | Father, asymptomatic AIP | c.669_698del | |
| 3 | 1 | F | IUGR | Mother, symptomatic AIP | c.669_698del |
| 4 | 1.5 | M | VACTERL association | Father, asymptomatic AIP | c.669_698del |
| 5 | 8 | F | Father, asymptomatic AIP | c.669_698del | |
| 6 | 12 | M | Father, symptomatic AIP | c.669_698del | |
| 7 | 9 | F | Sideroblastic anaemia and language delay | Father, asymptomatic AIP | c.669_698del |
| 8 | 7 | F | Language delay | Mother, symptomatic AIP | c.669_698del |
| 9 | 16 | M | Mother, asymptomatic AIP | c.76C>T |
Four cases (45%) presented with associated abnormalities. Cases 7 and 8 presented with language delay. Case 7 also suffered from sideroblastic anaemia caused by a homozygous c.683G>T mutation in the SLC25A38 gene. Case 5 had multiple congenital anomalies compatible with VACTERL association (imperforate anus with rectovesical fistula, lumbar hemivertebrae, superior vena cava draining into the coronary sinus, polycystic left kidney and left grade 5 vesicoureteral reflux).
Case 4 (history of intrauterine growth restriction [IUGR]; birth weight, 1630g below first percentile) had significant growth retardation at age 3 years (weight and height below first percentile).
The mother of case 4 was diagnosed during pregnancy. She had an acute attack during gestation and required intravenous administration of haem. The child was delivered by emergency C-section due to IUGR and maternal preeclampsia at 36 weeks gestation.
To conclude, the results of our study support the hypothesis that there is a high prevalence of AIP in children in our region due to the founder effect of the c.669_698del mutation: 28 cases per million inhabitants. All patients were asymptomatic at diagnosis and remained so during followup (with a possible unconfirmed attack in case 9), which may be due to the young age of the population under study (7 years). We found a high rate of associated abnormalities (language delay, multiple congenital anomalies, etc.) that had not been reported previously, so additional follow-up studies need to be conducted on larger samples. Attacks during pregnancy could be a risk factor for IUGR in the newborn and hypertension in the mother, but this aspect is still being debated.6
Early diagnosis is essential in order to implement measures to avoid attack-precipitating factors, to address the chronic problems caused by the disease, and to provide adequate counselling to the family.
We want to thank the patients and their families for their collaboration. This study was partially funded by the UCAM-Universidad Católica de Murcia (PMAFI/09/14). M. Barreda-Sánchez is a graduate student on a predoctoral fellowship awarded by the same university.
Please cite this article as: Sánchez-Soler MJ, Barreda-Sánchez M, Ballesta-Martínez MJ, Glóver G, Guillén-Navarro E. Porfiria aguda intermitente en población pediátrica de la región de Murcia: fenotipo y prevalencia. An Pediatr (Barc). 2016;84:114–115.
