






La pubertad precoz, a pesar de las definiciones clínicas estandarizadas y pruebas de diagnóstico disponibles, requiere, en ciertas situaciones una investigación exhaustiva y estructurada con el fin de conocer la causa.
Niña de 4 años de edad, fenotípicamente de sexo femenino, enviada a la consulta de endocrinología pediátrica por pubarquia y telarquia. Se observó aceleración en la tasa de crecimiento con niveles altos de estradiol y testosterona, con determinaciones prepúberes de la hormona luteinizante y foliculoestimulante. El resto del estudio de pubertad precoz periférica mostró la presencia de gonadoblastoma bilateral. El estudio genético reveló cariotipo 46 XY con mutación c.89G> T (p.Arg30Ile) en el exón 1 del gen SRY, confirmando el diagnóstico de disgenesia gonadal completa.
Los trastornos de la diferenciación sexual deben ser considerados en el abordaje y la investigación de las causas de la pubertad precoz periférica, especialmente en presencia de tumores de ovario, como gonadoblastoma y disgerminomas.
Despite standard clinical definitions and availability of diagnostic tests for precocious puberty, an intensive and structured investigation is needed in order to diagnose the aetiology in particular cases.
A 4-year-old, phenotypically female child was referred to paediatric endocrinology consultation for premature pubarche and thelarche. There was an acceleration of growth velocity with high levels of estradiol and testosterone, and prepubertal FSH and LH measurements. Investigation showed bilateral gonadoblastoma as the cause of the peripheral precocious puberty.
Genetic studies revealed 46 XY karyotype with mutation c.89G> T (p.Arg30Ile) in exon 1 of the SRY gene, confirming the diagnosis of complete gonadal dysgenesis. Disorders of sexual differentiation must be considered in the approach and investigation of peripheral precocious puberty, especially in the presence of ovarian tumours, such as gonadoblastoma and dysgerminoma.
La pubertad precoz (PP) por definición corresponde al desarrollo de los caracteres sexuales secundarios antes de los 8 años en las mujeres y de los 9 años en los varones1,2.
La etiología de PP es amplia, desde variantes de la normalidad, como telarquia precoz aislada, hasta el riesgo de enfermedades con comorbilidad y mortalidad significativas como tumores de células germinales.
El enfoque de la PP implica su clasificación en 2 subtipos: la pubertad precoz central (PPC), dependiente de gonadotropinas (causada por la maduración temprana del eje hipotálamo hipófiso gonadal [HHG]), y la pubertad precoz periférica (PPP), independiente de las gonadotropinas (debida a la secreción excesiva de hormonas sexuales, andrógenos o estrógenos derivados de las glándulas suprarrenales, de las gónadas o exógena)2.
La PPP puede ser de origen genético (testotoxicosis; hiperplasia suprarrenal congénita; mutación gen DAX1; síndrome de McCune-Albright) o adquirida (quiste ovárico; tumor ovárico, testicular, suprarrenal o productores de fracción β de gonadotropina coriónica humana [β-HCG]), esteroides sexuales exógenos)3.
La PPP adquirida se produce secundariamente a un aumento de esteroides sexuales exógenos o endógenos.
La evaluación diagnóstica incluye una anamnesis y un examen físico detallado y pruebas complementarias, como la determinación de gonadotropinas basales y tras estímulo con LHRH (esencial para el diagnóstico diferencial entre PPC y PPP), pruebas hormonales (testosterona, 17-β-estradiol, sulfato de dehidroepiandrosterona [DHEA-S], androstendiona y 17-hidroxiprogesterona [17-OH-progesterona], β-HCG, tiroxina libre [T4 libre] y hormona tiroestimulante [TSH]) y pruebas de imagen (radiografía de la mano-muñeca para determinar la edad ósea, ecografía pélvica o testicular, abdominal y RM craneal). Por último, y ante una fuerte sospecha clínica deben, realizarse estudios genéticos3.
Caso clínicoNiña enviada a la consulta de endocrinología pediátrica a los 4 años y 2 meses por sospecha de pubertad precoz.
Resultado de un embarazo a término con adecuada somatometría para la edad gestacional y sin antecedentes familiares, perinatales o patológicos importantes.
Presentaba un desarrollo psicomotor adecuado para la edad y para la evolución estaturo-ponderal, con peso por encima del percentil 95 y cruce en el percentil de la talla, variando entre el percentil 25-50 hasta los 2 años y luego valores por encima del percentil 95 a los 4 años (fig. 1).
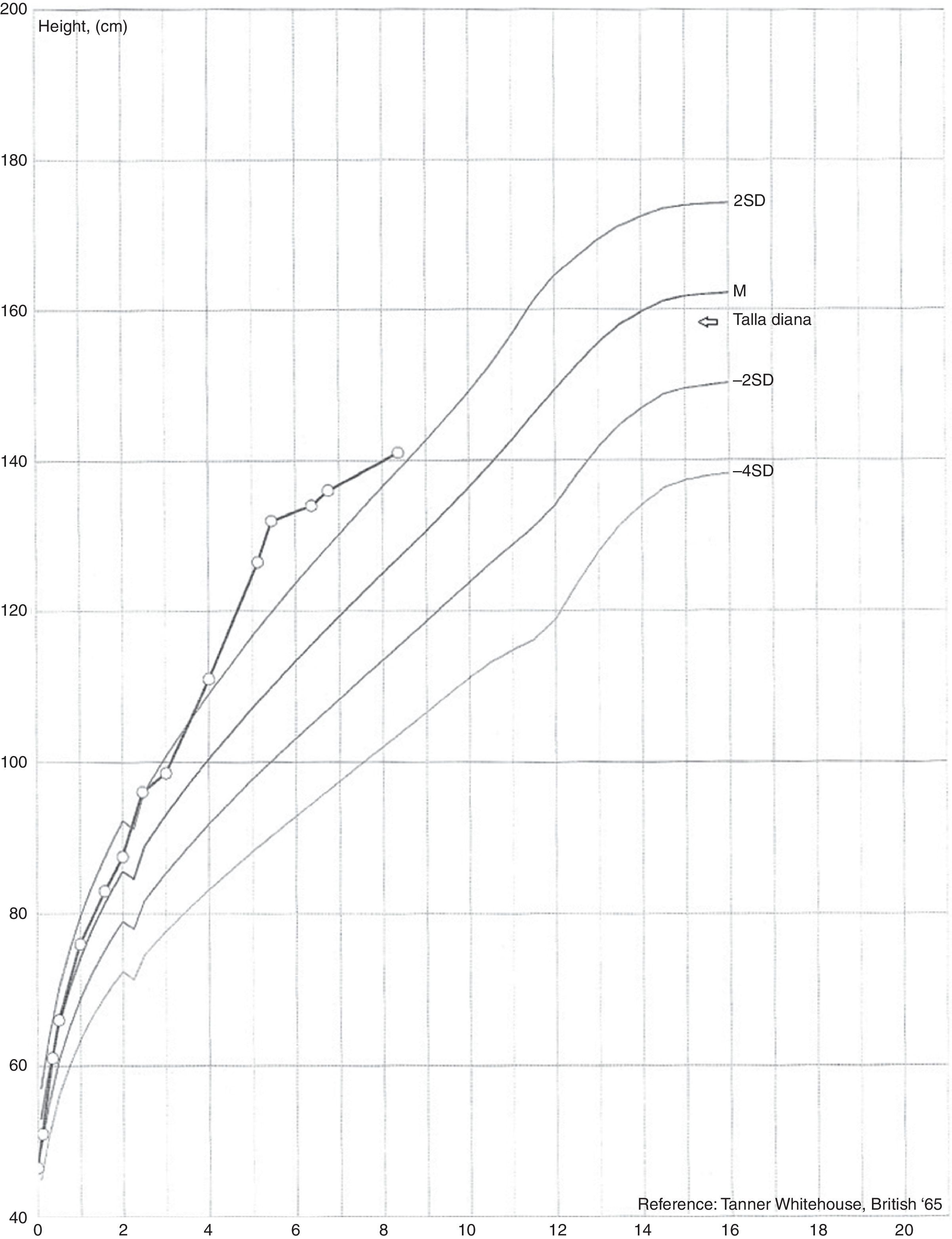
Se observó desarrollo de caracteres sexuales secundarios a los 4 años, con aparición de vello púbico y el brote de mama. En la primera evaluación hospitalaria, tenía un peso de 22kg (> P95), talla de 115,5cm (SDS 3,10), velocidad de crecimiento de 14,81cm/año (SDS 7,13), talla diana 159cm (SDS –0,50), genitales externos femeninos con estadio de Tanner A1-B2-P2.
El laboratorio de investigación reveló niveles elevados de estradiol, testosterona total y β-HCG (tabla 1).
Valores hormonales y marcadores tumorales
| Resultado | Valor de referencia | |
| 17-hidroxiprogesterona | 90ng/dl | 4-115ng/dl |
| Sulfato dehidroepiandrosterona | 27,1μg/dl | 5-35μg/dl |
| Delta-4-androstenediona | 0,357ng/ml | 0,85-2,75ng/ml |
| Tiroxina libre | 7,5μg/dl | 7,3-15μg/dl |
| Hormona tiroestimulante | 1,5μU/ml | 0,6-6,3μU/ml |
| IGF1 | 297ng/ml | 16-396ng/ml |
| Hormona adrenocorticotropa | 15,5pg/ml | 7-28pg/ml |
| Cortisol | 7μg/dl | 3-20μg/dl |
| Prolactina | 13,5ng/ml | 2,6-18ng/ml |
| Lactato deshidrogenasa | 257U/l | 110-295/l |
| Alfa-fetoproteína | 1,6ng/ml | 0-15ng/ml |
| B-HCG | 10,3U/l | ≤ 1U/l |
| FSH | 0,3/ml | < 5mU/ml |
| LH | < 0,1mU/l | < 5mU/l |
| Estradiol | 17,3pg/ml | < 12,5pg/ml |
| Testosterona total | 1,21ng/ml | 0,06-0,8ng/ml |
β-HCG: fracción beta de la hormona gonadotrofina coriónica humana; FSH: hormona foliculoestimulante; IGF1: factor de crecimiento insulínico tipo 1; LH: hormona luteinizante.
Los niveles de 17-OH-progesterona, DHEA-S, delta-4-androstenediona, T4 libre, TSH, hormona adrenocorticotropa, cortisol (por la mañana), prolactina, lactato deshidrogenasa (LDH) y alfafetoproteína (α-FP) fueron normales para la edad (tabla 1). En suero, FSH 0,3 mU/ml y LH < 0,1 mU/l, y la prueba de LH/RH reveló una respuesta prepuberal (pico de LH 1,1 mU/ml; pico de FSH 2,3 mU/ml, relación LH/FSH < 1).
La edad ósea (EO) estaba acelerada 19 meses en relación con la edad cronológica (EC) (EO: 5 años y 9 meses, EC: 4 años y 2 meses). La ecografía pélvica reveló una imagen circunscrita focal hiperecogénica con 25mm de diámetro en la región anexial izquierda. La tomografía computarizada pélvica reveló lesiones anexiales bilaterales sólidas, con matriz calcificada, de 30mm la derecha y 25mm la izquierda.
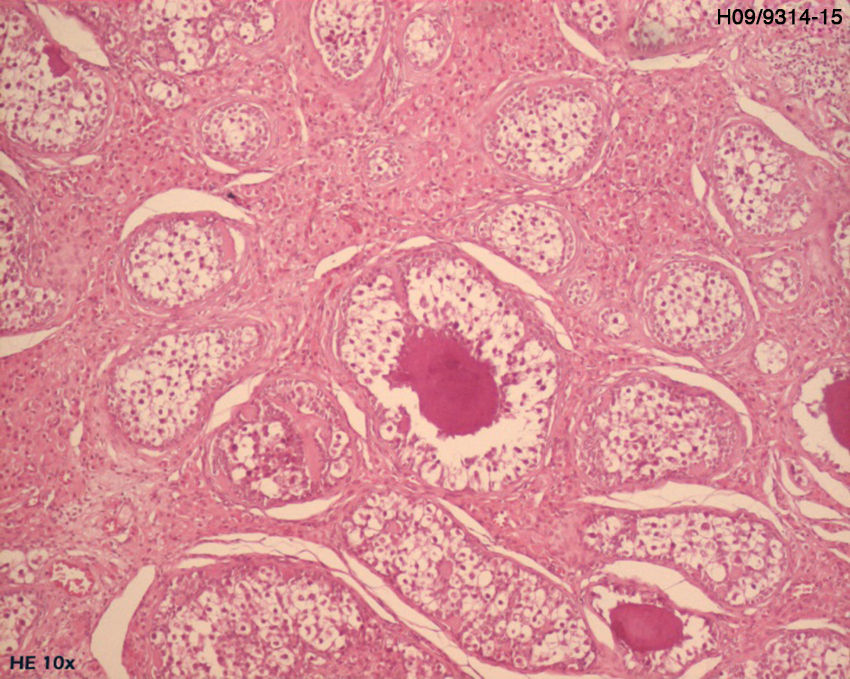
El examen histológico de la biopsia mostró parches tisulares ocupados por estructuras de gonadoblastoma bilateral, confirmada por inmunohistoquímica (CD117 e inhibina alfa).
Se sometió a una ooforectomía bilateral laparoscópica. El análisis anatomohistopatológico confirmó la presencia de un gonadoblastoma bilateral (figs. 2 y 3).

El estudio genético en sangre periférica reveló un cariotipo 46 XY y la mutación c.89G> T (p.Arg30Ile) en el exón 1 del gen SRY (sex–determining region of the Y chromosome), que confirma el diagnóstico de disgenesia gonadal completa XY por una mutación en el gen SRY.
DiscusiónLos gonadoblastomas son tumores benignos raros, compuestos por células germinales mixtas con nidos circunscritos de cordones de células sexuales, en general, con una membrana basal hialina y con calcificaciones difusas o focales4. Fue descrito por primera vez en 1953 por Scully5.
En el 50% de los casos hay un crecimiento excesivo y anormal de las células germinales, con progresión hacia disgerminoma y, ocasionalmente, otros tumores germinales, como el carcinoma embrionario y el coriocarcinoma6. Estos tumores son raramente causas de pubertad precoz7.
En este caso, la aparición de telarquia y pubarquia a los 4 años, con una marcada aceleración de la velocidad de crecimiento, era indicativa de una afección orgánica como causa del PP. El estudio de laboratorio reveló niveles elevados de estradiol y testosterona, con niveles prepuberales de FSH y LH. Estos hechos orientaron al estudio de causas periféricas. La evaluación pélvica identificó la presencia de masas ováricas que, asociadas a la elevación de β-HCG, plantearon el diagnóstico de afección tumoral.
La β-HCG es una hormona glucoproteica que, además de su asociación con el embarazo, se considera un marcador tumoral para el cáncer ginecológico, así como, más raramente, de tumores no ginecológicos8. Los tumores son activos desde el punto de vista hormonal, con capacidad para producir β-HCG9.
Otros posibles marcadores de células tumorales germinales, como la LDH y la α-FP, tuvieron valores dentro de la normalidad. También Capito et al., en un estudio realizado con 11 casos de disgenesia gonadal pura 46 XY, describieron, en un paciente de 17 años con desarrollo puberal incompleto para la edad, la presencia de gonadoblastoma y disgerminoma con niveles normales de α-FP10.
En el caso presentado, la biopsia confirmó una afección tumoral, identificándose gonadoblastoma bilateral.
Estos tumores se presentan casi exclusivamente en pacientes con disgenesia gonadal asociada a la presencia de un cromosoma Y, siendo los cariotipos frecuentes en estas situaciones 46 XY o mosaicismo 45X/46XY6.
La presentación clínica es variable, desde la ausencia de síntomas a diferentes grados de virilización o feminización y la presencia de masa abdominal11.
La presentación con signos de pubertad precoz parece estar relacionada con la secreción autónoma de esteroides sexuales, que se produce en el 15% de los casos, estando condicionados por componentes tumorales derivados de las células de los cordones sexuales. La secreción de andrógenos induce virilización o seudopubertad isosexual, si la secreción de estrógenos es dominante10.
Esta fuerte asociación de gonadoblastomas con trastornos de la diferenciación sexual condujo a la realización de un estudio genético en el caso presentado, confirmándose el diagnóstico de disgenesia gonadal completa 46 XY con identificación de la mutación c.89G> T (p.Arg30Ile) en el exón 1 del gen SRY. Esta mutación fue descrita por primera vez en 2002 por Assumpção et al. como causa de disgenesia gonadal12.
La disgenesia gonadal completa se caracteriza por cintillas gonadales, el desarrollo normal de los conductos de Müller y genitales externos femeninos10,13. Esta alteración de la diferenciación sexual es resultado de un fallo de desarrollo de los testículos fetales, secundario a mutaciones en ciertos genes, como SRY, SOX9, SF1, DAX1 y WT110,14. Las mutaciones del gen SRY representan solo el 10-20% de los trastornos de la diferenciación sexual 46 XY, ocurriendo en los restantes casos por mutaciones en los otros genes referidos13.
La ooforectomía sigue siendo el tratamiento de elección debido al alto riesgo de transformación maligna.6
Además de la ooforectomía, el tratamiento incluye terapia hormonal e inducción de la fertilidad, debido a la infertilidad de todos estos pacientes.
En conclusión, se hace hincapié en la importancia del conocimiento de la fisiología de la pubertad normal por los profesionales sanitarios, para ser capaces de reconocer las anomalías en el desarrollo puberal, cuya etiología puede condicionar gran morbimortalidad, como es el caso de los tumores ováricos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
