


La enfermedad de Castleman o hiperplasia angiofolicular linfoide es una entidad infrecuente en pediatría. Su etiología es desconocida; se cree que existe una estimulación antigénica crónica de probable origen viral1,2 (VHS-8, VIH, VEB) o alteración de la regulación de factores de crecimiento3,4 (IL-6, VEGF), originando proliferación anómala de células plasmáticas.
Histológicamente se distinguen tres formas:
- –
Hialino-vascular (HVV): hiperplasia folicular anómala con importante vascularización interfolicular. Es la más frecuente.
- –
Plasmo-celular (PVV): abundantes células plasmáticas y ausencia de proliferación vascular en el espacio interfolicular.
- –
Mixta.
Existen dos formas clínicas:
- –
Unicéntrica (UCD): más frecuente en niños con predominio femenino. Se suele presentar como hallazgo casual de adenopatía única de localización mediastínica, cervical y/o abdominal5. El 90% de los casos corresponde con forma hialino-vascular, siendo menos frecuente la plasmo-celular o la mixta,en cuyo caso pueden acompañarse de manifestaciones sistémicas (fiebre, pérdida de peso, sudoración o astenia). La cirugía es curativa, reservándose la radioterapia para los casos con mala respuesta6. Tiene buen pronóstico, salvo la forma plasmo-celular, que podría requerir tratamientos adicionales y su curso no es conocido.
- –
Multicéntrica (MCD): más frecuente en adultos de edad avanzada especialmente varones, excepcional en niños. La mayoría de los casos corresponden a la forma plasmo-celular. Se asocia a VHS-8 en pacientes VIH positivos y en el 40% de VIH negativos4. Suele presentarse con poliadenopatías, hepatoesplenomegalia y síntomas sistémicos inespecíficos. Son frecuentes las alteraciones analíticas (anemia, trombopenia, hipoalbuminemia, aumento de VSG).
Se han descrito asociaciones a amiloidosis, pénfigo vulgar y síndrome de POEMS7. Un tercio asocian enfermedades malignas (sarcoma de Kaposi y linfoma no Hodgkin).
Su tratamiento es discutido. Existen múltiples modalidades que incluyen esteroides a dosis altas, radioterapia, quimioterapia, antivirales (ganciclovir o foscarnet), inmunomoduladores (IFN-alfa) y más recientemente anticuerpos monoclonales frente a IL-6 (atlizumab) o frente CD 20 (rituximab)6-9. Presenta una mortalidad en la edad adulta del 50%. Existe menor morbimortalidad5 en niños.
En este sentido, presentamos un niño de 5 años remitido para estudio de poliadenopatías. En los últimos 6 meses presentó adenopatías múltiples cervicales, sin respuesta a tratamiento médico, por lo que precisa exéresis quirúrgica y estudio histológico.
Tras la intervención presenta fiebre elevada, astenia y palidez intensa, apreciándose una anemia aguda (Hb 5,4g/dl), que precisó de transfusión de hemoderivados. Se realizó una ecografía abdominal donde se detectaron múltiples adenopatías intraperitoneales e infiltración renal bilateral.
Antecedentes familiares: padre fallecido por carcinoma gástrico.
Antecedentes personales: abscesos cervicales por Mycobacterium avium, que requirieron drenaje quirúrgico y tratamiento con tuberculostáticos a los 2 años de edad.
Exploración física: destacaba puntos de sutura en región cervical izquierda por exéresis ganglionar y lesiones cicatriciales en la región cervical derecha. Soplo sistólico II/VI sin repercusión hemodinámica. Abdomen blando y depresible, sin masas ni visceromegalias.
Pruebas complementarias: anemia microcítica. VSG: 59mm/s. AFP: normal, colinesterasa 4.413 U/ml, enolasa 27,5 ng/ml y LDH 610 U/l. Hipoalbuminemia e hipergammaglobulinemia. Frotis de sangre periférica: linfocitos grandes con núcleos convolutos, citoplasma basófilo y granulación azurófila. Anisopoiquilocitosis con basofilia difusa de la serie roja y anisotrombocitosis.
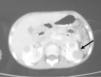
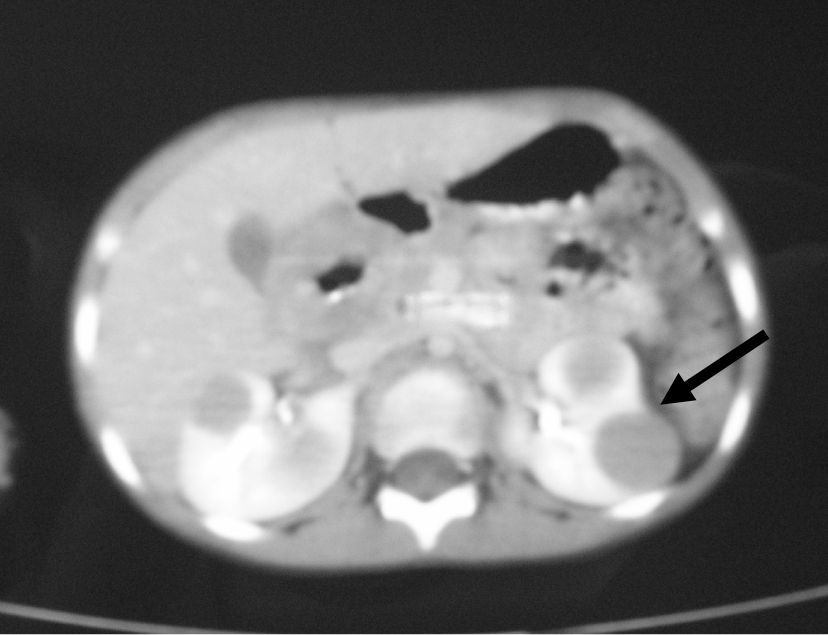
TC cervical y toraco-abdominal (fig. 1): múltiples adenopatías en la región cervical izquierda, con zonas de calcificación central y a nivel retroperitoneal con afectación renal bilateral. Radiografía de tórax: normal. Biopsia ganglionar cervical: enfermedad de Castleman; forma mixta con predominio hialino-vascular.
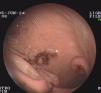
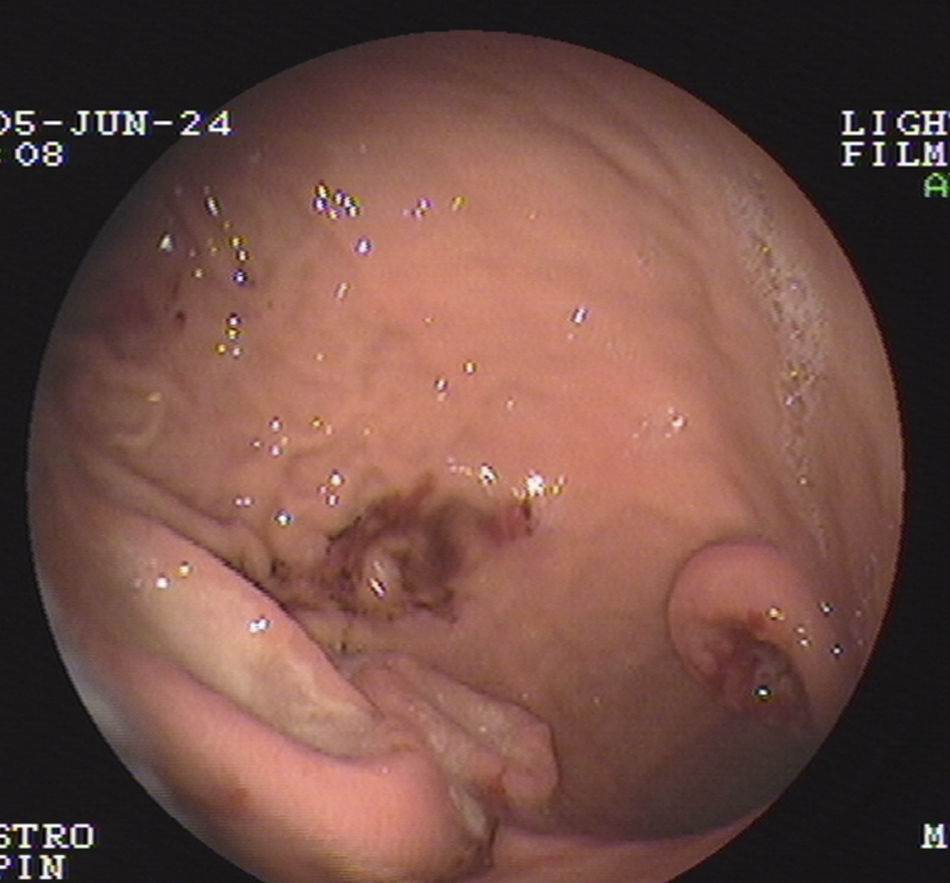
Durante su estudio, presentó dolor abdominal y melenas, por lo que se realizó una endoscopia gastroduodenal donde se visualiza la presencia de 3 tumoraciones con cráter central, ulcerado, leñoso, con bordes elevados rodeados de mucosa macroscópicamente normal (fig. 2). Estudio histológico: linfoma no Hodgkin tipo B gástrico. Inmunohistoquímica: CD-20 positivo. CD-3, CD-5, CD-10, CD-30, BCL-2 y Ki-67, negativos. Mapa óseo, citología del LCR y aspirado y biopsia de médula ósea, normales.
Se comenzó tratamiento con corticoides a dosis de 60mg/m2. Tras confirmación diagnóstica de linfoma no Hodking en estadio III, se inició quimioterapia según protocolo LMB 89 grupo B (COPADM) con desaparición de la sintomatología y normalización las alteraciones analíticas. Actualmente, asintomático y libre de enfermedad a los 36 meses de seguimiento.
La enfermedad de Castleman es una entidad infrecuente en pediatría. Se han descrito 86 casos con edades comprendidas entre los 2 meses y los 17 años, con predominio de sexo femenino.
La forma multicéntrica es excepcional en la edad pediátrica. Se suele corresponder con el tipo histológico plasmo-celular, siendo ocasional el mixto o hialino-vascular.
La asociación con linfoma no Hodgkin ocurre en un 20-25% de los casos en la forma multicéntrica. Son escasos los casos descritos en la literatura en niños.
La quimioterapia constituye una opción terapéutica en formas multicéntricas. El régimen más usado incluye ciclofosfamida, vincristina, prednisona y doxorrubicina, consiguiéndose tasas de remisión del 55%. A diferencia de los adultos, en niños mejor pronóstico.
Considerar a la enfermedad de Castleman en el diagnóstico diferencial de poliadenopatías por su asociación a enfermedad maligna e implicaciones pronósticas.
