




La lipodistrofia congénita generalizada es un trastorno hereditario poco común, de herencia autosómica recesiva, caracterizado por la ausencia casi total de tejido adiposo desde el nacimiento. Se asocia a la aparición precoz de anomalías metabólicas como hipertrigliceridemia, esteatosis hepática y resistencia a la insulina, que pueden acarrear consecuencias fatales debido al desarrollo de aterosclerosis precoz, diabetes lipoatrófica y cirrosis hepática. Los autores presentan el caso de un paciente diagnosticado clínica y analíticamente en el primer año de vida y, posteriormente, confirmado por la identificación de una mutación en el gen BSCL2. Con este caso los autores pretenden compartir las dificultades en el manejo terapéutico de la dislipidemia y la diabetes en esta enfermedad.
Congenital generalised lipodystrophy is a rare autosomal recessive disorder characterised by a marked deficiency of adipose tissue and usually recognised at birth. This disorder is associated with early development of metabolic complications such as hypertriglyceridemia, hepatic steatosis, and insulin resistance. These complications ultimately lead to fatal events as a consequence of early atherosclerosis, lipoatrophic diabetes and hepatic cirrhosis. The authors report the case of a patient diagnosed, based on clinical and laboratory findings, in the first year of life. The established diagnosis was then confirmed by identifying a mutation in the BSCL2 gene. Because the hypertriglyceridemia and diabetes were refractory to treatment, the authors present this case in order to reflect on the best therapeutic management of this pathology.
La lipodistrofia congénita generalizada (LCG) fue descrita inicialmente por Berardinelli1 en Brasil, el año 1954, y confirmada en 1959 por Seip2 en Noruega, dando origen a la denominación de lipodistrofia congénita de Berardinelli-Seip. Se han reportado aproximadamente 250 casos de diferentes etnias, estimándose una prevalencia mundial de 1:10.000.000 de habitantes3.
La LCG es un trastorno hereditario con transmisión autosómica recesiva. Fueron identificados varios genes implicados en la enfermedad, clasificándola en subtipos. La LCG tipo 1 es causada por mutaciones del gen AGPAT2, localizado en el cromosoma 9q34, y que codifica una enzima 1-acilglicerol-3-fosfato-O-aciltransferasa 2 implicada en la biosíntesis de fosfolípidos y triglicéridos4. La LCG tipo 2 se debe a una mutación en el gen BSCL2, localizado en el cromosoma 11q13, y que codifica la proteína seipina, cuya función se desconoce5. Recientemente han sido descritas la LCG tipo 3, con mutaciones del gen CAV1, y la LCG tipo 4, causada por mutaciones del gen PTRF. Ambos genes codifican factores determinantes en la homeostasis lipídica y en la captación celular de glucosa dependiente de insulina6,7.
La principal característica de la enfermedad es la ausencia casi total de tejido adiposo y consiguiente apariencia muscular generalizada desde el nacimiento3. Después, durante la infancia, surge hipertrigliceridemia, con posible aparición de xantomas3. Entre los 8 y 10 años aparece la resistencia a la insulina con acantosis nigricans marcada3. La diabetes generalmente se instala después de los 12 años8. Pueden encontrarse diversas anomalías hepáticas que van desde hepatomegalia, ligero aumento de las transaminasas, esteatosis hepática y más tarde cirrosis8.
Con este caso los autores pretenden compartir las dificultades en el manejo terapéutico de la dislipidemia y la diabetes en esta enfermedad.
Observación clínicaSegundo hijo de padres jóvenes, de raza caucásica, procedentes de la región de Minho (Portugal), sanos y consanguíneos de primer grado. Embarazo controlado, a término, sin incidencias. Parto eutócico y con somatometría adecuada para la edad gestacional. Período neonatal sin incidencias.
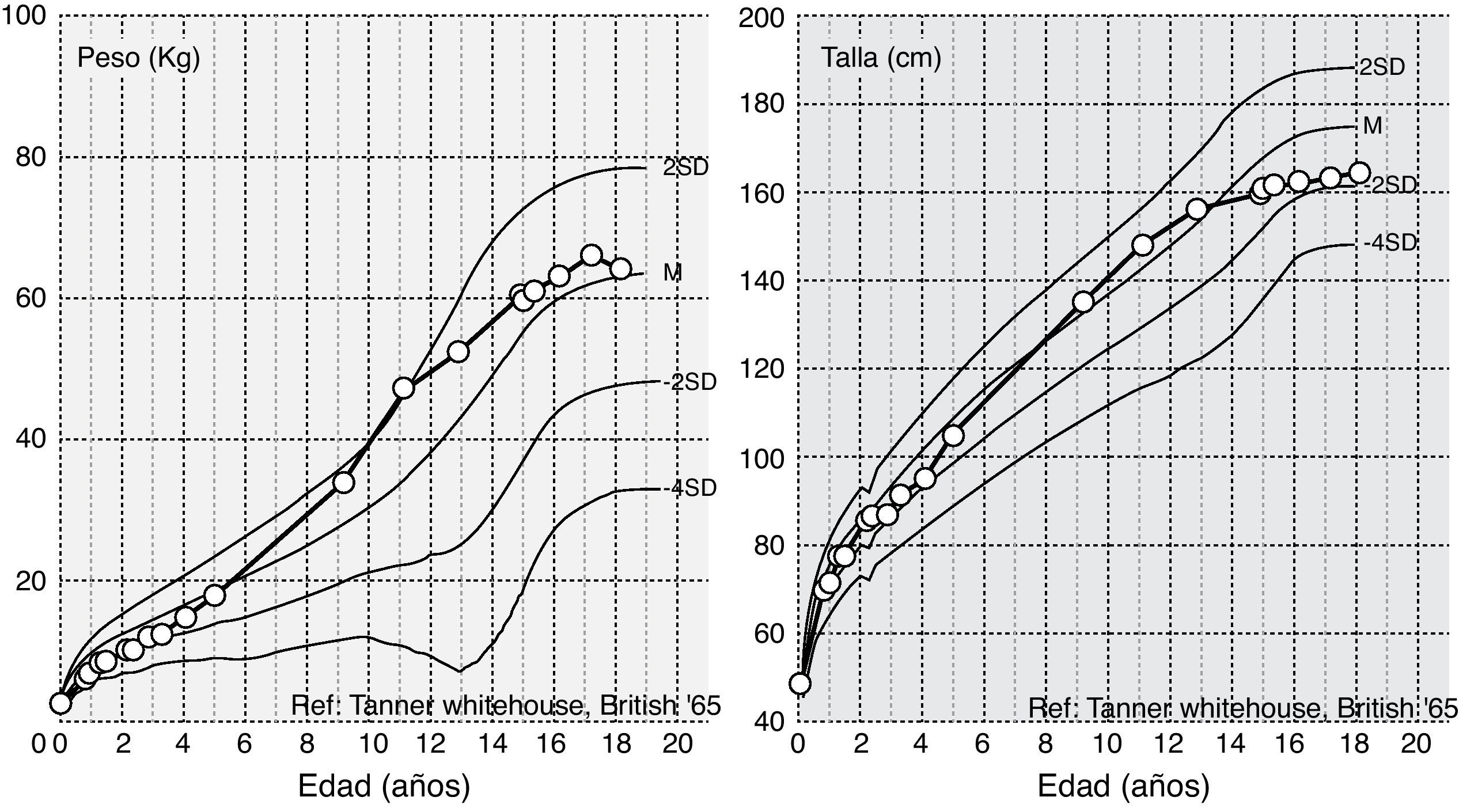
El diagnóstico de LCG se realizó durante el primer año de vida. En los 3 primeros años de vida, a pesar de tener un apetito voraz, se observó una desaceleración del peso. Desde entonces evolución dentro de la normalidad de peso y estatura (fig. 1). Al analizar los antecedentes patológicos se encontró hipertrigliceridemia (807mg/dl) a los 11 meses, esteatosis hepática, resistencia a la insulina y diabetes lipoatrófica a los 9, 11 y 14 años, respectivamente. Durante este período no se inició ningún tratamiento.
En edad escolar, por dificultades de aprendizaje, fue llevada a cabo una evaluación cognitiva que reveló cociente intelectual global de 68.
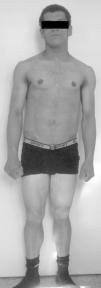
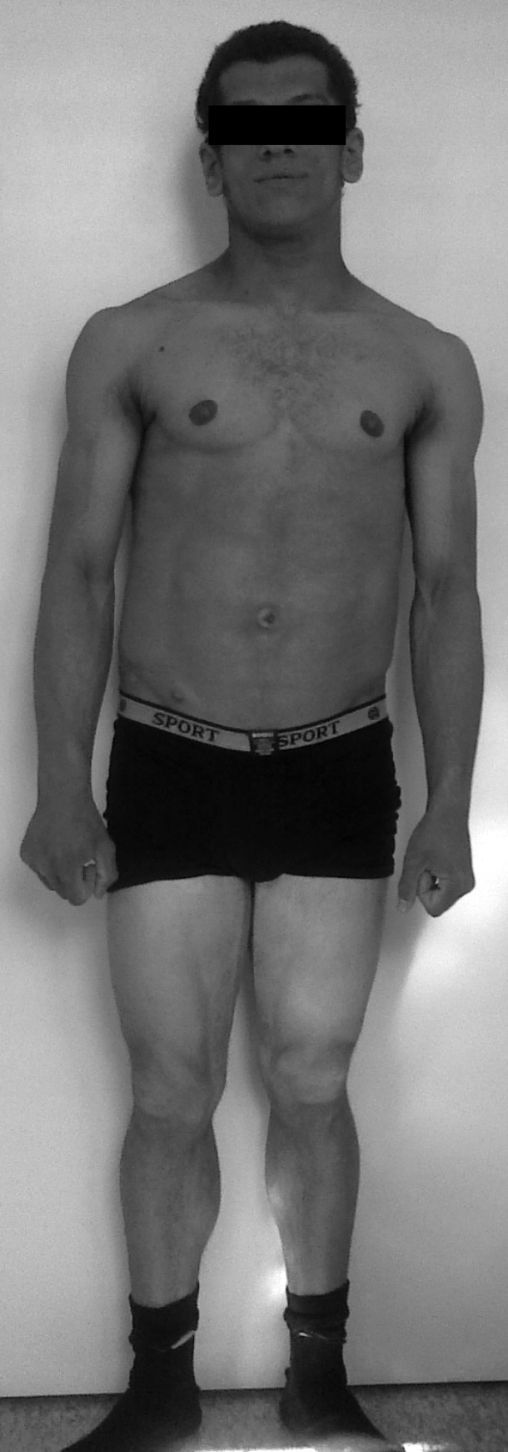
Fue referenciado a los 15 años a la unidad de endocrinología pediátrica. La exploración física reveló tensión arterial normal, peso 57 kg (percentil 50), estatura 161,4cm (percentil 21), índice de masa corporal 21,9 kg/m2 (percentil 83), apariencia muscular generalizada por ausencia casi total de tejido adiposo (fig. 2), acantosis nigricans exuberante en la región cervical y axilar, pequeños xantomas en las axilas, prognatismo, ombligo prominente y hepatomegalia de 4cm por debajo del reborde costal.
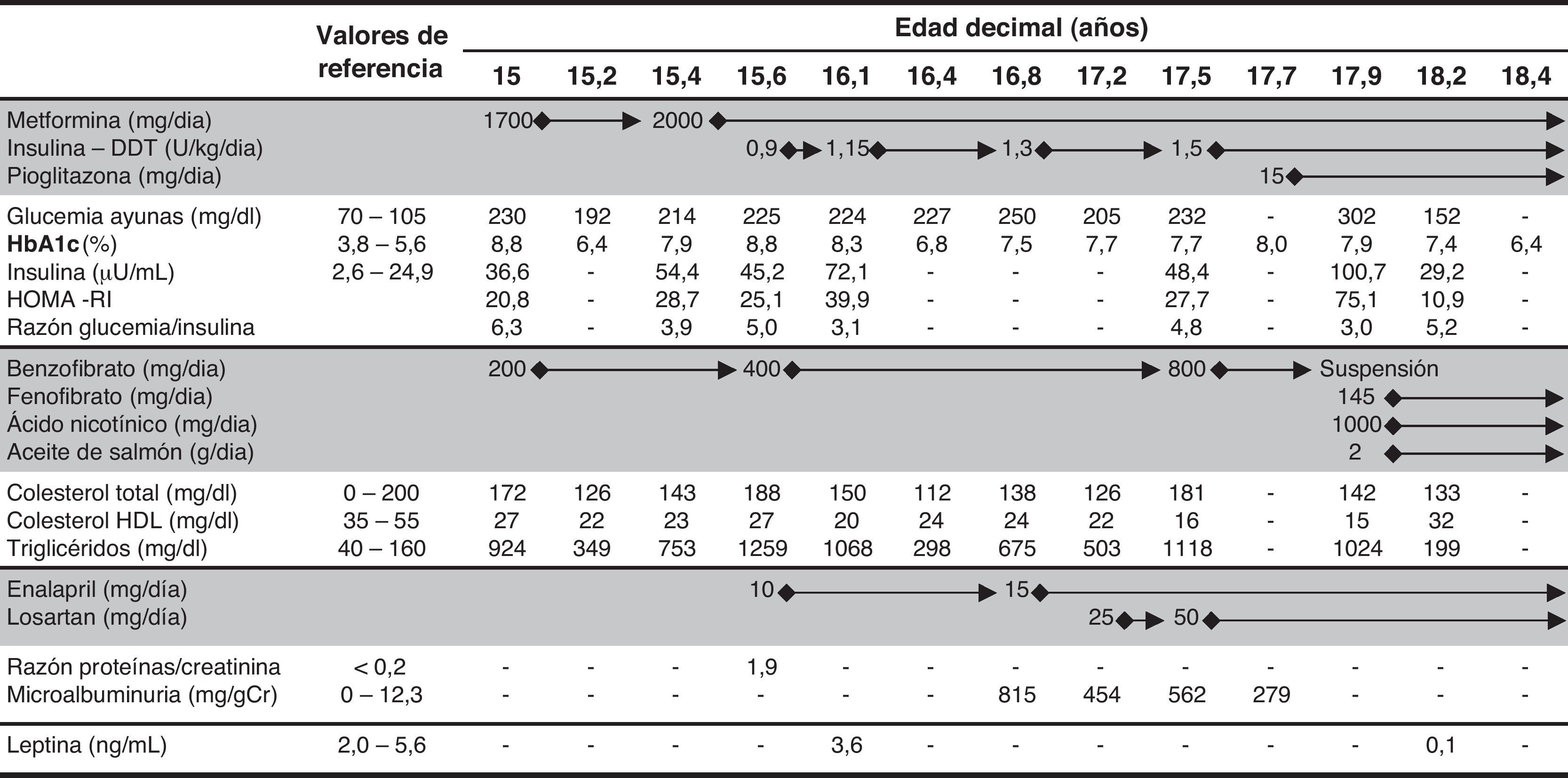
En ayunas presentaba los siguientes valores: glucemia 230mg/dl, insulina 36,6μU/ml, hemoglobina glucosilada 8,8%, triglicéridos 924mg/dl, colesterol total 172mg/dl, colesterol HDL 27mg/dl, AST 43 U/l y ALP 75 U/l (tabla 1). La ecografía abdominal reveló hepatomegalia de 20cm, con hiperecogenicidad difusa, y el ecocardiograma hipertrofia ventricular izquierda ligera.
El estudio de genética molecular, al identificar la mutación c.604C>T (p.Arg202X) en homocigosis en el exón 4 del gen BSCL2, confirmó el diagnóstico clínico de LCG y permite clasificarla como del tipo 2.
La tabla 1 muestra la evolución analítica con el tratamiento establecido.
Para el tratamiento de la diabetes se eligió inicialmente metformina en monoterapia. Después hubo necesidad de asociar insulinoterapia (régimen basal-bolus), con aumento gradual de la dosis diaria total (dosis máxima 1,5 U/kg/día). Más tarde también empezó pioglitazona. A los 15,6 años se detectó proteinuria, iniciando terapéutica con enalapril. Aproximadamente un año después fue necesario aumentar la dosis por microalbuminuria significativa. Posteriormente se añadió losartán por respuesta insuficiente.
En cuanto a la hipertrigliceridemia fue aconsejada dieta hipolipídica y hipocalórica, incentivada la práctica de ejercicio físico e iniciado benzofibrato. A pesar del aumento de dosis no se logró el control de la hipertrigliceridemia, por lo que dicha terapéutica fue sustituida por la asociación de aceite de salmón, fenofibrato y ácido nicotínico, con buena respuesta.
Aunque inicialmente la leptina sérica fuera normal (3,6 ng/ml), recientemente se observó hipoleptinemia (0,1 ng/ml). Los valores séricos de factor de crecimiento semejante a la insulina tipo 1 siempre fueron normales.
DiscusiónEl paciente descrito presenta diversas manifestaciones clínicas que sugieren LCG. La principal característica fenotípica es la ausencia casi total de tejido adiposo presente desde el nacimiento. Sin embargo es poco frecuente establecer el diagnóstico en el período neonatal9. Generalmente, este aspecto típico sólo es valorado más tarde, cuando se asocian otras características. En este caso, la valoración se produjo durante la investigación de la desaceleración en el peso y de la hipertrigliceridemia que mientras tanto se instaló. Con frecuencia estos pacientes, durante los primeros años de vida, no muestran un aumento de peso de acuerdo con su apetito voraz, probablemente debido a un hipermetabolismo8.
Actualmente es posible diferenciar genéticamente los distintos subtipos de LCG, correspondiendo el caso presentado al tipo 2, el más frecuente en Portugal10. Clínicamente, este subtipo en comparación con el tipo 1 tiene mayor prevalencia de déficit cognitivo y miocardiopatía hipertrófica10,11. El paciente descrito presenta discapacidad cognitiva ligera y hipertrofia ventricular izquierda.
A nivel metabólico presenta la historia natural típica de LCG, con hipertrigliceridemia grave y resistencia a la insulina, que con el tiempo originó diabetes. La base fisiopatológica de estos trastornos no se conoce totalmente, pero parece basarse en la ausencia de adipocitos con capacidad de metabolismo y almacenamiento lipídico. Por consiguiente, existe exceso de lípidos circulantes, especialmente triglicéridos y ácidos grasos libres, que se depositan en otros tejidos como el hígado y los músculos excediendo su capacidad oxidativa y de almacenamiento. Clínicamente hay agravamiento de la hipertrigliceridemia, lo que contribuye al desarrollo de esteatosis hepática y resistencia a la insulina. Por otro lado, la ausencia de tejido adiposo origina deficiencia de leptina, que también puede desempeñar un papel fundamental en la fisiopatología de estos trastornos3,8.
El control metabólico en estos pacientes implica un auténtico reto terapéutico, y se basa en reducir la hipertrigliceridemia, mejorar la resistencia a la insulina y controlar la diabetes. Los pacientes deben ingerir dieta hipocalórica y hipolipídica, con objeto de evitar la quilomicronemia. Deben cumplir un programa regular de ejercicio dinámico, para mejorar la sensibilidad a la insulina y la dislipidemia. Si aún así persistieran niveles elevados de triglicéridos se deberá iniciar terapéutica con fibratos y dosis altas de aceite de pescado, con ácidos grasos omega n-3 poliinsaturados3.
Para el control de la hiperglucemia se utiliza metformina en monoterapia o asociada a insulinoterapia. La metformina reduce la hiperinsulinemia, aumenta la sensibilidad a la insulina, reduce el apetito y mejora la esteatosis hepática8. Con la progresión de la enfermedad es necesario asociar insulinoterapia, con frecuencia en dosis altas a consecuencia de la extrema resistencia a la insulina. En este caso también se optó por iniciar pioglitazona. Este fármaco se une y activa el factor de transcripción nuclear peroxisome proliferator-activated receptory (PPARY), que es un regulador de la adipogénesis. Aunque la información sobre este fármaco en pacientes con LCG sea escasa en la literatura médica, Vitoria et al. presentaron un caso de LCG tipo 2 tratado con rosiglitazona, donde se comprobó una mejoría del control glucémico, de la sensibilidad a la insulina, así como un aumento de la leptina sérica12. Las oscilaciones analíticas observadas en nuestro paciente no se explican únicamente por la medicación utilizada, ya que la adhesión a la dieta y el cumplimiento de la insulinoterapia no siempre es fácil en adolescentes.
A pesar de que las recomendaciones de la International Society Pediatric and Adolescent Diabetes sobre el tratamiento de la microalbuminuria en pacientes diabéticos impliquen el uso de inhibidores de la enzima conversora de la angiotensina o bloqueantes de receptores AT1 de angiotensina II en monoterapia, en este caso se optó por una combinación de ambos, dada la escasa respuesta observada13. Aunque esta asociación sea objeto de controversia, algunos estudios sugieren que el bloqueo dual puede tener un mayor efecto antiproteinúrico que la monoterapia14.
Dado el importante papel de la leptina en la fisiopatología de los trastornos metabólicos que se encuentran en la LCG, Beltrand et al. examinaron el efecto de la leptina recombinante humana en 7 pacientes con LCG. En un primer trabajo, tras 4 meses de tratamiento, observaron una mejora en la reducción de los niveles séricos de triglicéridos, en la sensibilidad a la insulina y en el volumen hepático (un 63, un 30 y un 20,3%, respectivamente)15. Sin embargo, el mismo grupo publicó recientemente otro trabajo según el cual, tras 28 meses de tratamiento, la mayoría de los pacientes desarrolló resistencia total o parcial a los efectos metabólicos de la terapéutica, en parte explicado por el desarrollo de anticuerpos antileptina16.
Se trata de pacientes complejos cuyo control metabólico sigue siendo un reto, en los que la sensibilización para el cumplimiento de la dieta y de una terapéutica múltiple es esencial, en un intento de mejorar su esperanza de vida.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
