
La hipofisitis linfocítica (HL) es un proceso inflamatorio crónico de la glándula hipofisaria con probable base autoinmunitaria y amplio espectro clínico, pudiéndose afectar hipófisis y/o tallo hipofisario1–3 produciendo déficits hormonales variables adeno y/o neurohipofisarios4. Esta entidad es rara en la edad pediátrica, habiéndose comunicado 19 casos en menores de 14 años5–7. Las características clínicas, manejo y evolución a largo plazo son poco conocidos. El diagnóstico definitivo se basa en estudios histológicos, pero se acepta el diagnóstico de presunción basado en criterios clínicos específicos2. El tratamiento es controvertido, siendo aceptados el abordaje médico, quirúrgico o el conservador mediante seguimiento estrecho con resonancia magnética (RM) craneal2. En el caso que se presenta destacan la evolución autolimitada con un seguimiento a 10 años.
Niña de 9 años que consulta por poliuria y polidipsia bruscas (5-6 l/día) de 2 meses de evolución, sin pérdida de peso, alteración subjetiva del crecimiento, cefalea, alteraciones visuales, galactorrea ni otra sintomatología. Exploración física, incluido fondo de ojo, normal con talla 134cm (–0,7 DE), IMC 19,8kg/m2 (0,3 DE), estadio puberal 1 Tanner y talla diana 162,5cm (–0,2 DE). Estudio analítico: marcadores tumorales sanguíneos y anticuerpos-antihipófisis negativos; función hipofisaria con déficit de hormona antidiurética en el test de restricción hídrica, respuesta patológica de hormona crecimiento (GH) en 2 tests farmacológicos de estímulo (test de hipoglucemia insulínica y ornitina con picos de 2,6 y 2,2ng/ml, respectivamente), IGF1 233,9ng/ml (VN=117-771) e IGFBP3 1,75ng/ml (VN=1,58-3,99), sin otros déficits hormonales; líquido cefalorraquídeo con bioquímica, citología y marcadores tumorales normales. Estudio radiológico: radiografía lateral craneal y gammagrafía ósea sin alteraciones; RM con aumento del volumen adenohipofisario, engrosamiento y marcado realce del infundíbulo y ausencia de señal hiperintensa de neurohipófisis (fig. 1). Con el diagnóstico de presunción de infundíbulo-hipofisitis-linfocítica (INHL) se realizó seguimiento clínico estrecho con tratamiento específico para la diabetes insípida (DI) y controles periódicos de RM craneal para evaluar la necesidad de completar estudio con biopsia. A los 3 meses se detectó mejoría radiológica, con normalización del tamaño de la adenohipófisis, discreto engrosamiento del tallo y persistencia de ausencia de la señal normal neurohipofisaria (fig. 2.1 y 2.2). El déficit bioquímico de GH revirtió en el test de estímulo realizado a los 6 meses, siendo el crecimiento e inicio y posterior desarrollo puberal normales, con talla adulta 164cm (0 DE), menarquia a los 126/12 años y posteriores reglas regulares. La imagen patológica de hipófisis y tallo se normalizó al año de la seguimiento y 10 años después se mantiene normal, permaneciendo únicamente la ausencia de señal neurohipofisaria (fig. 2.2). Sigue precisando tratamiento con vasopresina para controlar la DI.
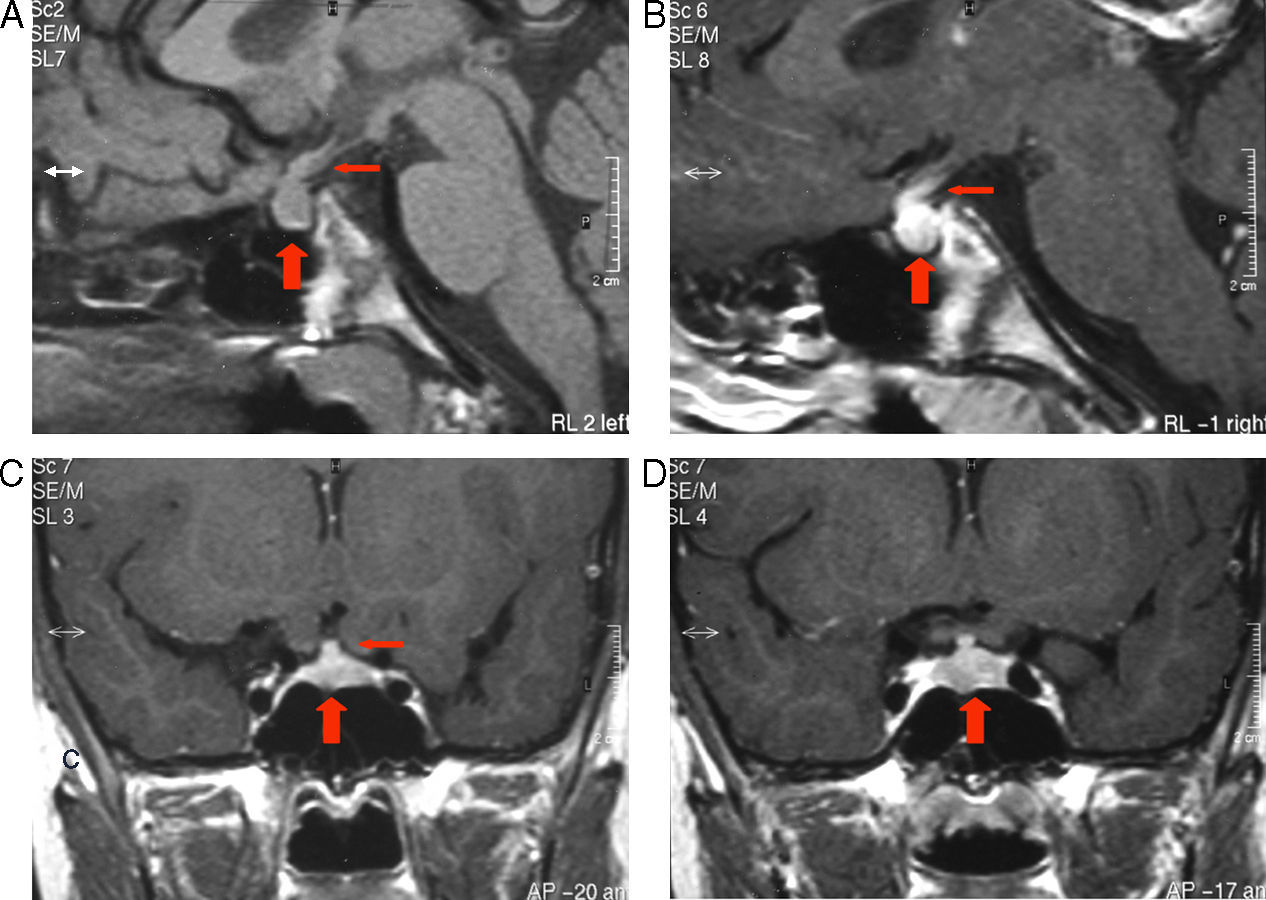
RM de región sellar al diagnóstico. A) Corte sagital sin contraste: ausencia de la señal normal de la neurohipófisis, aumento del volumen de la adenohipófisis (flecha gruesa) y engrosamiento del infundíbulo (flecha fina). B) Corte sagital con contraste: importante realce en infundíbulo (flecha fina) y tuber cinereum. C) Corte coronal con contraste: heterogeneidad de señal de la adenohipófisis (flecha gruesa) asociada a llamativo engrosamiento del tallo pituitario (flecha fina). D) Corte coronal con contraste: realce heterogéneo en la adenohipófisis (flecha gruesa).
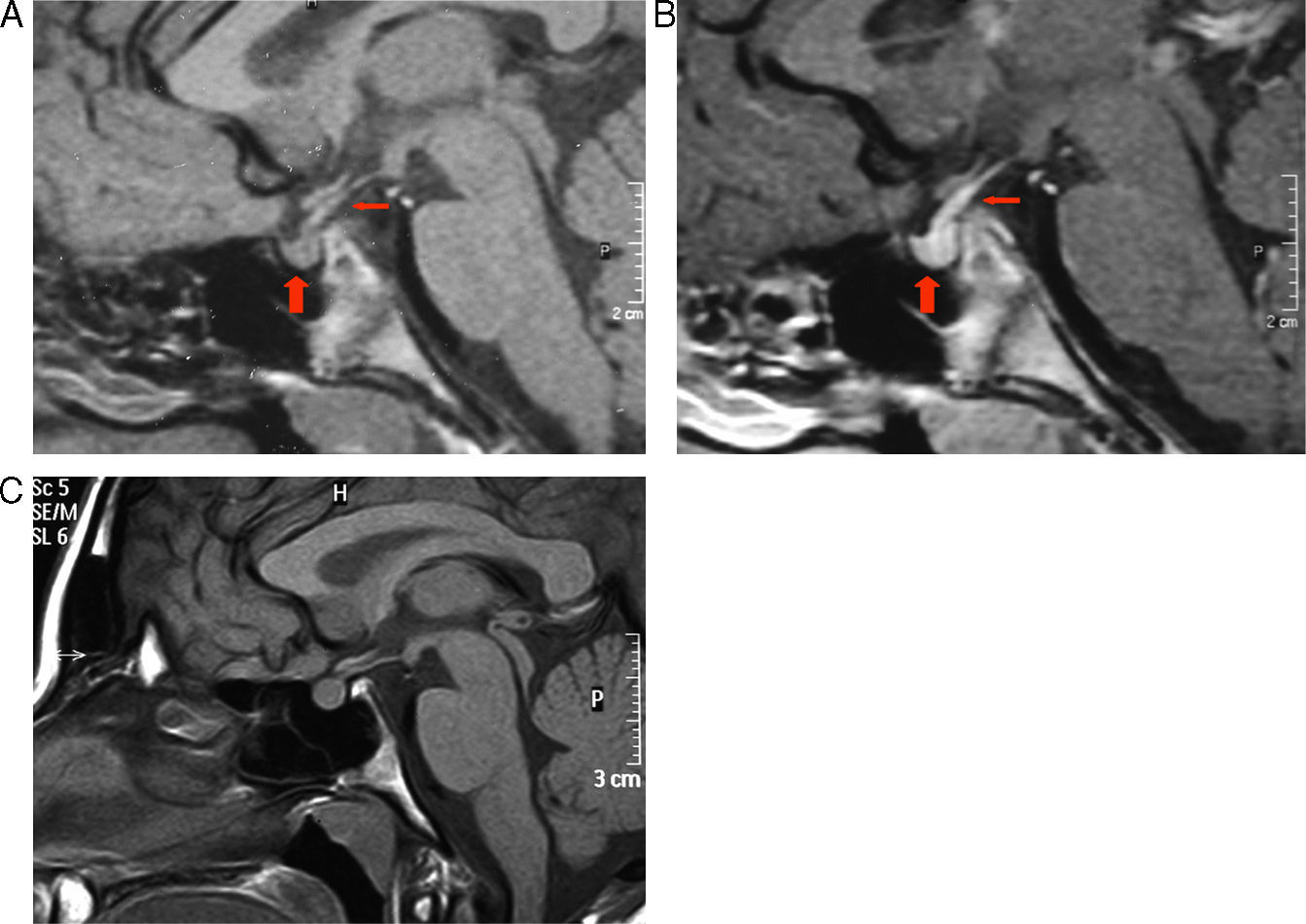
2.1) RM a los 3 meses del diagnóstico. 1A) Corte sagital sin contraste: normalización del tamaño de la adenohipófisis (flecha gruesa), persistencia de ausencia de la señal normal de la neurohipófisis y discreto engrosamiento del tallo pituitario (flecha fina). 1B) Corte sagital con contraste: realce normal de la adenohipófisis (flecha gruesa) y mayor del esperado en infundíbulo patológico (flecha fina). 2.2) RM a los 10 años. C) Corte sagital sin contraste: normalidad del tamaño de la adenohipófisis y tallo pituitario con ausencia de la señal normal de la neurohipófisis.
El seguimiento de nuestra paciente con INHL es el más largo hasta ahora reportado en menores de 14 años, con evolución de 10 años frente a una media de 2,9. Los datos clínicos, analíticos y los hallazgos radiológicos fueron acordes con el diagnóstico de INHL con afectación del tallo, adeno y neurohipófisis, siendo esta la presentación más común en niños5–7. Tras excluir otras lesiones infundíbulo-hipofisarias no adenomatosas (tuberculosis, histiocitosis X, infecciones, tumores) se adoptó una actitud conservadora con seguimiento clínico-radiológico como propugnan algunos autores en casos poco sintomáticos y con posibilidad de control estrecho2,3. Aunque la biopsia transesfenoidal es el «gold standard» para el diagnóstico, cada vez más autores sugieren que debe reservarse para pacientes con efecto masa grave, compresión del nervio óptico o deterioro clínico y/o radiológico progresivo2–4. La determinación de anticuerpos-antihipófisis con las técnicas actuales carece de suficiente sensibilidad/especificidad3,7, debiéndose interpretar con cautela. Guttenberg et al.8 encontraron anticuerposantihipófisis, específicos frente GH, en una paciente diagnosticada inicialmente de INHL; tras 2 meses de corticoterapia sin respuesta, una segunda biopsia evidenció un germinoma intrasellar. Por tanto, se precisarían nuevos antígenos más específicos3.
La historia natural de la HL es variable, evolucionando a fibrosis-atrofia glandular o, como nuestro caso, resolverse espontáneamente. La regresión es más frecuente en las formas de INHL, aunque en ellas la DI es permanente como en nuestra paciente2,5,7. Los resultados con corticoterapia son variables1,2,9,10. También se han utilizado agentes inmunosupresores11. La radioterapia estereotáxica ha sido beneficiosa en casos con gran efecto masa2,3. Independientemente del tratamiento, es necesario un seguimiento clínico estrecho12, realizando RM periódicamente para descartar complicaciones, como extensión de la masa o tumores ocultos, fundamentalmente germinomas en niños (20% de los casos descritos en menores de 14 años)5–7.
Aunque la biopsia sigue siendo el «gold standard» para el diagnóstico de HL, y dado que la utilidad de los anticuerpos antihipofisarios es limitada, de manera individualizada el diagnóstico puede basarse en los hallazgos clínico-radiológicos junto con la evolución y seguimiento. El uso sistemático de corticoides no está justificado. Es importante un seguimiento estrecho y prolongado para descartar la presencia de germinomas, inicialmente difíciles de diferenciar de la HL.
