


El síndrome de Sturge-Weber es un síndrome neurocutáneo raro cuya gravedad está determinada por el grado de afectación cerebral y el control de la epilepsia.
Los autores describen y analizan las características clínicas y de neuroimagen de este síndrome, a través de un estudio retrospectivo de 13 pacientes (8 niñas; con edad en la primera consulta entre los 15 días y los 9 años). Doce presentaban angioma facial y una atriquia en la zona de afectación cerebral. Se diagnosticó epilepsia en 6 casos, hemiplejía en 4, retraso psicomotor en 7 y glaucoma en 4 casos. Se encontraron alteraciones cerebrales en 10 niños, 3 sin sintomatología neurológica.
La clínica presentada es variable y no siempre hay relación entre la gravedad clínica y las anomalías de neuroimagen, que pueden aparecer hasta en ausencia de síntomas neurológicos.
Sturge Weber Syndrome is a rare neurocutaneous syndrome in which the severity is determined by degree of brain involvement and control of epilepsy. The authors describe and analyse clinical and imaging features of this syndrome, through a retrospective study of 13 patients (8 girls; aged between 15 days and 9 years at first visit). Twelve had facial angioma and one had atrichia corresponding to the area of brain involvement. Epilepsy was diagnosed in 6 cases, hemiplegia in 4, psychomotor delay in 7, and glaucoma in 4. Cerebral abnormalities were found in 10 children, 3 without neurological symptoms.
The clinical signs and symptoms vary and there is not always a relationship between the severity of the clinical and neuroimaging abnormalities, which may occur even in the absence of neurological symptoms.
El síndrome de Sturge-Weber (SSW) es un síndrome neurocutáneo raro (incidencia de 1/20-50.000) que se caracteriza por angiomas en el territorio inervado por la primera rama trigeminal, leptomeninges y/o coroides ocular1-4. La gravedad está determinada por el grado de afectación cerebral y el control de la epilepsia1-4.
El objetivo de este trabajo es describir y analizar las características clínicas y de neuroimagen de este síndrome.
Pacientes y métodosEstudio retrospectivo y descriptivo, basado en la consulta de los expedientes clínicos de una muestra consecutiva de 13 pacientes con diagnóstico de SSW seguidos en una consulta de neuropediatría. Se incluyó a pacientes con los 3 tipos de SSW reconocidos: angioma facial y leptomeníngeo con o sin glaucoma (tipo I), angioma facial y glaucoma (tipo II) y angioma leptomeníngeo sin angioma facial (tipo III)5-7. También se han incluido casos de angioma facial asociado a otro tipo de anomalías cerebrales típicas del SSW (tales como dilatación venosa profunda), anomalía venosa del cerebelo y retraso psicomotor. Se analizaron los siguientes parámetros: sexo, edad, manifestaciones clínicas y anomalías de neuroimagen.
ResultadosSe observó a 13 pacientes (8 niñas), con mediana de edad en la primera consulta de 4 meses (mínimo 15 días, máximo 9 años).
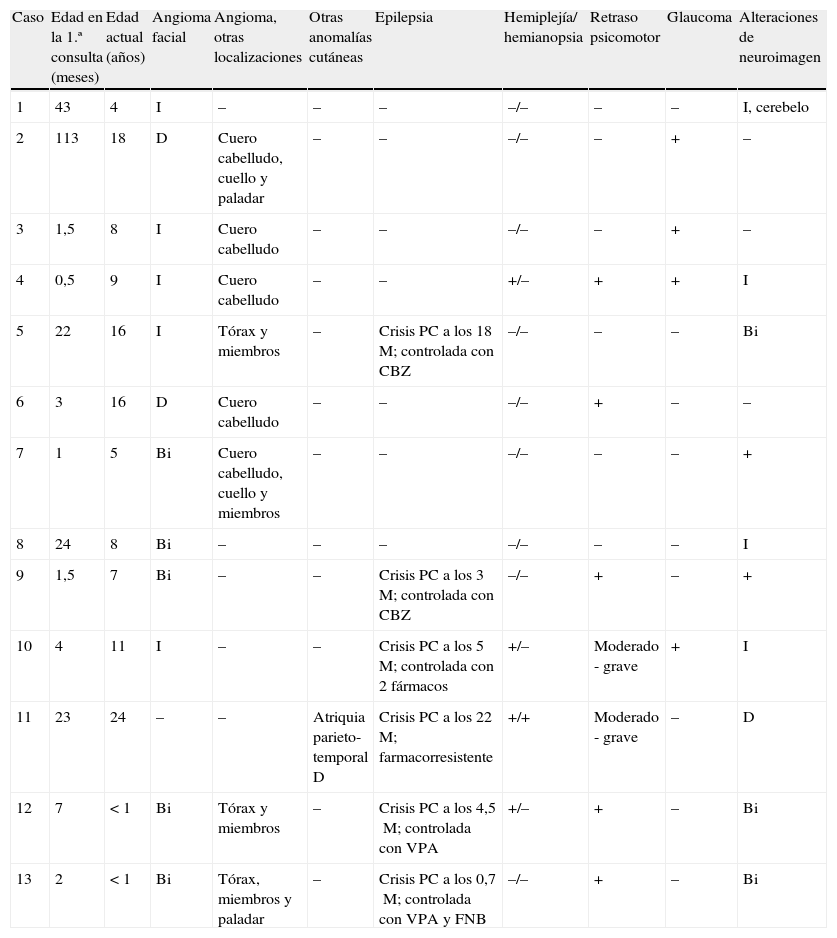
Doce niños presentaban el típico angioma facial, bilateral en 5 casos (fig. 1 y tabla 1). En 8 se observaron angiomas en otras localizaciones, como en los miembros en 4 (fig. 1 y tabla 1). La única paciente sin angioma facial presentaba una zona de atriquia en la región parieto-temporal derecha, correspondiente a la zona de afectación cerebral (fig. 1, caso 11).

Ejemplos de manifestaciones cutáneas: caso 4, angioma facial a la izquierda; caso 5, angiomas en la cara, el tronco y los miembros, de tonalidad más clara 8 años después; caso 10, angioma a la izquierda, más discreto 10 años después; caso 11, zona de atriquia ipsolateral a las lesiones cerebrales; caso 12, angiomas bilaterales en la cara, el tronco y los miembros.
Clínica presentada y alteraciones de neuroimagen
| Caso | Edad en la 1.ª consulta (meses) | Edad actual (años) | Angioma facial | Angioma, otras localizaciones | Otras anomalías cutáneas | Epilepsia | Hemiplejía/ hemianopsia | Retraso psicomotor | Glaucoma | Alteraciones de neuroimagen |
| 1 | 43 | 4 | I | – | – | – | –/– | – | – | I, cerebelo |
| 2 | 113 | 18 | D | Cuero cabelludo, cuello y paladar | – | – | –/– | – | + | – |
| 3 | 1,5 | 8 | I | Cuero cabelludo | – | – | –/– | – | + | – |
| 4 | 0,5 | 9 | I | Cuero cabelludo | – | – | +/– | + | + | I |
| 5 | 22 | 16 | I | Tórax y miembros | – | Crisis PC a los 18M; controlada con CBZ | –/– | – | – | Bi |
| 6 | 3 | 16 | D | Cuero cabelludo | – | – | –/– | + | – | – |
| 7 | 1 | 5 | Bi | Cuero cabelludo, cuello y miembros | – | – | –/– | – | – | + |
| 8 | 24 | 8 | Bi | – | – | – | –/– | – | – | I |
| 9 | 1,5 | 7 | Bi | – | – | Crisis PC a los 3M; controlada con CBZ | –/– | + | – | + |
| 10 | 4 | 11 | I | – | – | Crisis PC a los 5M; controlada con 2 fármacos | +/– | Moderado - grave | + | I |
| 11 | 23 | 24 | – | – | Atriquia parieto-temporal D | Crisis PC a los 22M; farmacorresistente | +/+ | Moderado - grave | – | D |
| 12 | 7 | < 1 | Bi | Tórax y miembros | – | Crisis PC a los 4,5M; controlada con VPA | +/– | + | – | Bi |
| 13 | 2 | < 1 | Bi | Tórax, miembros y paladar | – | Crisis PC a los 0,7M; controlada con VPA y FNB | –/– | + | – | Bi |
Bi: bilateral; CBZ: carbamazepina; D: a la derecha; FNB: fenobarbital; I: a la izquierda; M: meses; PC: parciales complejas; VPA: valproato.
Seis niños presentaron crisis epilépticas parciales complejas (edad de inicio: promedio de 9 meses, máximo 22 meses, mínimo 3 semanas). Cinco casos están controlados con medicación anticomicial (tabla 1). La única paciente con epilepsia farmacorresistente no tiene angioma facial y aguarda tratamiento quirúrgico (caso 11). Cuatro niños tienen hemiplejía, 1 hemianopsia y 7 retraso psicomotor, 2 de ellos sin epilepsia (tabla 1).
En 4 casos se asoció glaucoma, ninguno con pérdida total de la visión.
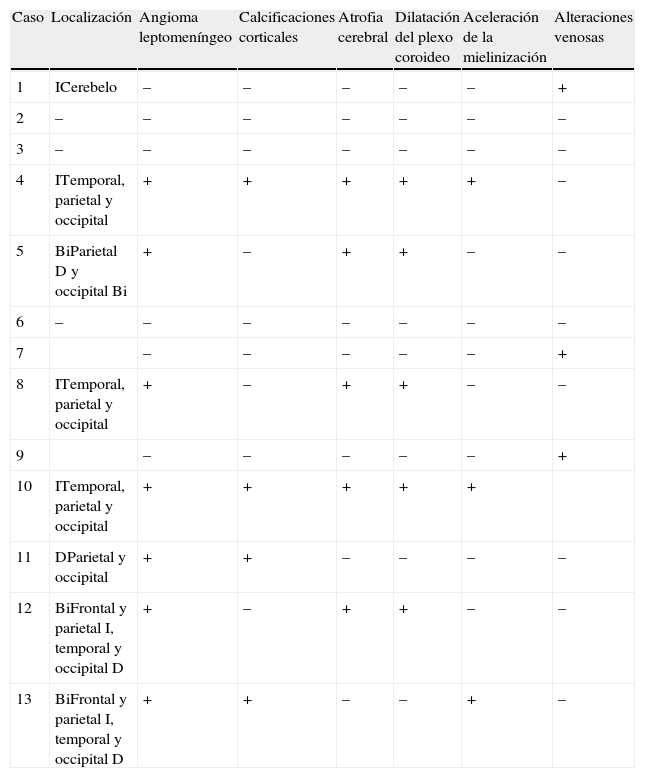
En 10 niños se encontraron alteraciones en la resonancia magnética (RM) cerebral: angioma leptomeníngeo (7), calcificaciones corticales (4), atrofia cerebral (5), dilatación del plexo coroideo (5) y/o alteraciones venosas: 2 dilatación venosa profunda, 1 malformación venosa en el cerebelo (fig. 2 y tabla 2). Dos de ellos no presentaban síntomas neurológicos (tabla 1).
 Caso 1, RM, corte axial, secuencia en T1 con contraste: angioma de la coroides a la izquierda (flecha). B) Caso 4, RM, corte coronal, secuencia en T2: calcificaciones corticales y atrofia parietooccipital a la izquierda (flecha). C) Caso 4, RM, corte axial, secuencia en T1 con contraste: angiomatosis pial (flecha) y atrofia cortical a la izquierda, con asimetría hemisférica y de la calota craneal. D) Caso 4, RM, corte axial, secuencia en T1 con contraste: angiomatosis pial, angioma del plexo coroideo (flecha) y atrofia cortical a la izquierda. E) Caso 10, RM, corte axial, secuencia T2: calcificaciones y atrofia del hemisferio izquierdo, gliosis del parénquima adyacente (flecha); dilatación de vena prepontina. F) Caso 10, RM, corte axial, secuencia en T2: calcificaciones y atrofia parietal a la izquierda; plexo coroideo mayor a la izquierda (flecha). G) Caso 11, RM, corte axial, secuencia en T1, con contraste: área encefaloclástica cortico-subcortical parieto-occipital, angiomatosis pial y plexo venoso más prominente a la derecha (flecha). H) Caso 11, TC, corte axial, sin contraste: calcificaciones corticales parieto-occipitales a la derecha (flecha).")
Ejemplos de anomalías de neuroimagen. A) Caso 1, RM, corte axial, secuencia en T1 con contraste: angioma de la coroides a la izquierda (flecha). B) Caso 4, RM, corte coronal, secuencia en T2: calcificaciones corticales y atrofia parietooccipital a la izquierda (flecha). C) Caso 4, RM, corte axial, secuencia en T1 con contraste: angiomatosis pial (flecha) y atrofia cortical a la izquierda, con asimetría hemisférica y de la calota craneal. D) Caso 4, RM, corte axial, secuencia en T1 con contraste: angiomatosis pial, angioma del plexo coroideo (flecha) y atrofia cortical a la izquierda. E) Caso 10, RM, corte axial, secuencia T2: calcificaciones y atrofia del hemisferio izquierdo, gliosis del parénquima adyacente (flecha); dilatación de vena prepontina. F) Caso 10, RM, corte axial, secuencia en T2: calcificaciones y atrofia parietal a la izquierda; plexo coroideo mayor a la izquierda (flecha). G) Caso 11, RM, corte axial, secuencia en T1, con contraste: área encefaloclástica cortico-subcortical parieto-occipital, angiomatosis pial y plexo venoso más prominente a la derecha (flecha). H) Caso 11, TC, corte axial, sin contraste: calcificaciones corticales parieto-occipitales a la derecha (flecha).
Alteraciones de neuroimagen
| Caso | Localización | Angioma leptomeníngeo | Calcificaciones corticales | Atrofia cerebral | Dilatación del plexo coroideo | Aceleración de la mielinización | Alteraciones venosas |
| 1 | ICerebelo | – | – | – | – | – | + |
| 2 | – | – | – | – | – | – | – |
| 3 | – | – | – | – | – | – | – |
| 4 | ITemporal, parietal y occipital | + | + | + | + | + | – |
| 5 | BiParietal D y occipital Bi | + | – | + | + | – | – |
| 6 | – | – | – | – | – | – | – |
| 7 | – | – | – | – | – | + | |
| 8 | ITemporal, parietal y occipital | + | – | + | + | – | – |
| 9 | – | – | – | – | – | + | |
| 10 | ITemporal, parietal y occipital | + | + | + | + | + | |
| 11 | DParietal y occipital | + | + | – | – | – | – |
| 12 | BiFrontal y parietal I, temporal y occipital D | + | – | + | + | – | – |
| 13 | BiFrontal y parietal I, temporal y occipital D | + | + | – | – | + | – |
Bi: bilateral; D: a la derecha; I: a la izquierda.
La clínica fue variable; sin embargo, el típico angioma facial con afectación de la región frontal y/o el párpado superior fue un signo prácticamente constante, siendo el principal indicador del diagnóstico. Un niño con angioma facial en esta localización tiene un riesgo del 10-25% de tener lesiones cerebrales y/o oculares concomitantes, lo que parece tener relación con la extensión de los angiomas faciales, especialmente si son bilaterales1,5. No obstante, se observó que un paciente con angioma facial bilateral presentaba solo dilatación venosa profunda en la RM, sin síntomas neurológicos (caso 7). Además, el caso más grave desde el punto de vista neurológico no presentaba angioma facial (caso 11). Esta paciente tenía una forma de presentación cutánea rara: atriquia en la zona de afectación cerebral. Está descrito por lo menos un caso similar8. Los autores consideraron que se trata de una variante de SSW, ya que cerca del 13% de los casos no tienen el angioma facial3,5,9-14.
Cuatro pacientes presentaban angiomas en los miembros, una localización característica del síndrome de Klippel-Trenaunay, estando ya descrita la ocurrencia de fenotipos sobrepuestos entre los dos síndromes2,5.
Manifestaciones ocularesLos angiomas oculares conducen a la aparición de glaucoma, la complicación ocular más frecuente que aparece en el 30-40% de los casos5 y que se observó en el 31% de la muestra. Si no se trata, puede causar pérdida de visión1,2, lo que no sucedió en ningún caso. A pesar de que su aparición es más frecuente durante el primer año de vida, puede aparecer a cualquier edad1,2. Por eso, todos los niños con SSW deben ser vigilados regularmente por un oftalmólogo.
Manifestaciones neurológicasLos vasos cerebrales anómalos condicionan alteraciones en el tejido cerebral circundante5. La isquemia cortical crónica evoluciona hacia áreas de calcificación, gliosis o atrofia cerebral, pudiendo provocar la aparición de síntomas neurológicos15.
Las crisis epilépticas se presentan en el 80% de los pacientes y ocurren habitualmente antes de los 2 años de edad1,5,16. Las primeras crisis suelen ser parciales, pero a lo largo del tiempo evolucionan a crisis generalizadas5,17. Más raramente pueden manifestarse como espasmos en flexión, mioclonías o crisis atónicas2,5,17,18. De los 13 pacientes estudiados, solo 6 (46%) presentaban epilepsia, que se manifestó en todos como crisis parciales complejas antes de los 2 años de edad. En cerca del 40% de los casos hay buena respuesta al tratamiento con antiepilépticos3, tal y como se observó en 5 de los 6 niños con epilepsia. Crisis frecuentes y prolongadas provocan un agravamiento de las lesiones cerebrales. La aparición de epilepsia antes del año de edad, así como la mala respuesta al tratamiento antiepiléptico son, por tanto, factores de mal pronóstico2,5,19. Se debe considerar la lobectomía/hemisferectomía en los casos de epilepsia farmacorresistente, como en el caso 1120.
Cerca del 33% de los pacientes presentan hemiplejía, tal como se observó en 4 niños de la muestra (31%), debido a la oclusión venosa o anoxia de los tejidos locales. Habitualmente es transitoria, pero la recurrencia de los episodios evoluciona a una hemiplejía persistente1,2,5.
La hemianopsia se detecta en cerca del 40% de los pacientes y aparece cuando hay compromiso del lóbulo occipital, lo que sucedió en el caso 113.
Cerca del 60% de los pacientes tienen retraso del desarrollo psicomotor, que parece tener relación con la extensión de las lesiones cerebrales y con la gravedad y la edad de inicio de la epilepsia1,2,5,19. En el grupo analizado, 7 pacientes (54%) presentan retraso del desarrollo, aunque 2 sin epilepsia y 2 sin lesiones cerebrales en la RM (tabla 1).
Imagen cerebralLa RM cerebral es el examen de elección para evaluar el grado de afectación cerebral. La imagen más típica es el angioma leptomeníngeo, que se traduce por captación de contraste en la duramadre en las secuencias T1, y que se observó en casi la mitad de los casos2. Otras imágenes típicas son la dilatación del plexo coroideo, áreas de calcificación y atrofia corticales, presentes en casi la mitad de la muestra, y dilatación del sistema venoso profundo, presente en 2 casos15,17.
Las lesiones cerebrales son ipsolaterales al angioma facial y en la mayoría, unilaterales. El compromiso bilateral ocurre en el 7-26% y se asocia a un mayor riesgo de presentar epilepsia refractaria y retraso psicomotor1,15. Tres pacientes de la muestra tenían lesiones cerebrales bilaterales, todos con epilepsia, y 2 de ellos con retraso del desarrollo.
En la RM cerebral de una paciente se observó una anomalía venosa de localización atípica en el cerebelo (caso 1). Ya que no había afectación arterial o anomalía estructural cerebelosa o de otro tipo concomitante, se descartó el diagnóstico de síndrome PHACE y se consideró el diagnóstico de SSW. Están descritos muy pocos casos de SSW con anomalías cerebelosas, todos ellos también con afectación supratentorial4,6,15,17.
Frente a un niño con angioma facial en el territorio inervado por la primera rama trigeminal, se debe realizar RM cerebral siempre que surjan síntomas neurológicos o a los 2 años de edad en los pacientes asintomáticos1,5. En edades precoces puede no haber anomalías de neuroimagen; así, si la RM es normal en los lactantes sintomáticos, es necesario repetirla con 1-2 años de edad1,5. Por otro lado, puede haber alteraciones de neuroimagen sin síntomas neurológicos, como en 2 niños de esta muestra, por lo que se debe realizar RM incluso en los niños asintomáticos. Si la RM es normal a los 2 años de edad, no es necesario repetirla, a menos que aparezcan síntomas1,5.
La tomografía computarizada permite la visualización de áreas de calcificación cortical (fig. 2), pudiendo tener alguna utilidad sólo cuando la RM no está disponible5.
En resumen, la clínica presentada es variable y no siempre hay relación entre la gravedad de las manifestaciones cutáneas, neurológicas y oculares entre sí, y con las alteraciones cerebrales, que pueden ocurrir hasta en ausencia de síntomas neurológicos.
Conflicto de interesesLos autores declaran que no existe ningún conflicto de intereses.
