

La progeria es un síndrome de envejecimiento precoz. El síndrome de Werner (SW) es un tipo de progeria del adulto que cursa con cataratas juveniles bilaterales y cambios esclerodermiformes cutáneos. Se produce a causa de la mutación del gen WRN que codifica una helicasa, enzima de reparación del ADN.
Se presenta el caso de una paciente femenina de 12 años con características de SW, pero sin mutación identificable del gen WRN, por lo que se clasifica como SW atípico.
Progeria is a premature ageing syndrome. Werner Syndrome (WS) is a type of progeria in the adult which includes bilateral juvenile cataracts and cutaneous sclerodermiform changes; it is caused by a mutation if the WRN gene which codes a helicase, a DNA repair enzyme.
A case is presented of a patient, a 12 year old girl, with characteristics of WS but with no identifiable mutation in the WRN gene, therefore it was classified as atypical Werner Syndrome (AWS).
En los seres humanos aparecen casos esporádicos de síndromes que tienen características de vejez prematura o acelerada. Son, en apariencia, defectos genéticos pleiotrópicos que se designan con el nombre de progeria.
Se han descrito 2 tipos de progeria: infantil y del adulto. La forma infantil es la del síndrome de Hutchinson–Gilford y la forma adulta es la del síndrome de Werner (SW), que reproduce con mayor fidelidad los cambios asociados con el envejecimiento y el patrón de enfermedades. Este trastorno fue el tema de tesis doctoral de Otto Werner en 1904. Es una enfermedad autosómica recesiva producida por la mutación de un gen que codifica para una enzima de reparación del ADN una helicasa ubicado en el cromosoma 8p12–p11.2. El diagnóstico es clínico y suele observarse en la cuarta década de la vida. Los pacientes suelen presentar cataratas juveniles bilaterales, baja estatura, obesidad troncular, pérdida de la grasa subcutánea, cambios esclerodermiformes en la piel y atrofia cutánea que condicionan la llamada «cara de pájaro». Se han publicado más de 400 casos, pero la mayor incidencia de la enfermedad se presenta en Japón donde existe un alto grado de consanguinidad.
El SW atípico (SWA) es otra forma de síndrome progeroide. Los pacientes presentan signos de envejecimiento precoz con algunos fenotipos diferentes a los pacientes con SW. El 15% de estos pacientes atípicos tiene mutaciones del gen lamina A/C (LMNA)1, el restante no tiene mutación identificable y se considera secundario a un defecto desconocido en el gen que codifica helicasa.
CasoMujer de 12 años de edad, que acudió a la consulta por presentar las mismas características físicas de la madre y un informe de patología que indicaba que estaba afectada de esclerodermia.
Antecedentes familiares: madre con iguales características. Falleció a la edad de 36 años a causa de un cáncer ovárico. No había consanguinidad.
Antecedentes personales: el embarazo fue complicado a causa de capacidad uterina disminuida, múltiples amenazas de parto pretérmino y desnutrición. Hubo parto pretérmino y el nacimiento fue por cesárea debido a las condiciones maternas; no hay disponible antecedentes perinatales. Recibió alimentación con leche de fórmula desde el nacimiento porque la madre presentó atrofia de la glándula mamaria. El desarrollo psicomotor estaba dentro de los parámetros normales, aunque actualmente presenta leves deficiencias pragmáticas y deficiencias motoras influenciadas por sus características físicas en la piel.
Examen físico: su peso era de 30kg P/E (cm T/E (
Inspección general: los segmentos corporales eran desproporcionados, la talla era baja, presentaba obesidad troncular y los miembros inferiores y superiores estaban hipotróficos.
Craneofacial: se observó exoftalmos leve, nariz puntiaguda, desarrollo dental irregular con protrusión de la arcada dentaria; «cara de pájaro». No presentaba cataratas y tenía voz ronca.
Abdomen: se observó un abdomen prominente con hepatomegalia de 2cm aproximadamente.
Genitales: femeninos, sin desarrollo puberal. Presentaba estadio 1 en la escala de Tanner.
Extremidades: delgadas, con manos y pies pequeños; los pies eran planos.
Piel: seca, acartonada, con poca elasticidad, abundante pilificación e hiperpigmentada, con cambios esclerodermiformes.
Ver características fenotípicas en la figura 1.

Estudios realizados: se realizó una biopsia de la piel con diámetro de 0,2×0,2cm y profundidad de 0,3cm. Espesor completo. Se observó tejido colágeno en la epidermis y en la hipodermis. La paciente presentaba esclerodermia.
Cariotipo con nivel de bandas GTG 700: la constitución cromosómica era 46 XX.
Citogenética: marcada fragilidad cromosómica.
Perfil tiroideo: normal. La TSH fue de 1,2658μIU/ml (VR: 0,3500–4,9400μIU/ml) y la T4 libre fue de 1,21ng/dl (0,70–1,48ng/dl).
Ecografía abdominal: se observó hígado graso.
Colesterol total: 227mg/dl (VN: <200mg/dl).
Glucemia basal: 93mg/dl.
Ecocardiograma Doppler color modo M y bidimensional: normal.
Densitometría ósea: presentó osteopenia.
TAC craneofacial simple: presentó alteraciones del desarrollo de los huesos nasales y de la espina nasal anterior, con protrusión del maxilar superior.
Por la presencia de dismorfismo, los cambios característicos en la piel y los patrones de enfermedad asociada se planteó la impresión diagnóstica de progeria, motivo por el que el genetista la valoró y consideró probable caso de progeria (SW), dado los hallazgos clínicos y del cariotipo cromosómico con bandeo que reveló alteraciones citogenéticas.
ResultadosSe contactó al International Registry of Werner Syndrome, dirigido por George Martin y Junko Oshima, ubicado en el departamento de enfermedad de la Universidad de Washington (Seattle), donde se enviaron las muestras de sangre para su análisis y se realizó la búsqueda de la región codificante del gen causante del SW (el gen WRN).
Los estudios moleculares no mostraron mutaciones del gen WRN. Los investigadores concluyen que el 90% de los pacientes con SW tienen una mutación detectable y el 10% restante todavía no tienen un cambio discernible en este locus. De igual manera no se detectó mutación del gen LMNA causante del síndrome progeroide atípico en pacientes sin mutación del gen WRN. Por lo tanto, con este análisis no se descarta completamente la posibilidad y se plantea como SWA o como síndrome progeroide atípico, debido a que clínicamente posee el fenotipo.
DiscusiónLa progeria es un síndrome de envejecimiento precoz. Todas estas condiciones son muy raras en la población y, por lo tanto, en muchos casos no hay casuística suficiente para realizar estudios estadísticos significativos.
Alrededor de 1.300 casos se han notificado en todo el mundo desde el año 1916 hasta el 2002, de los que cerca de 1.000 pacientes son japoneses2.
El SW es un tipo de progeria del adulto caracterizado por la mutación del gen WRN. Los expertos no creen que el gen encontrado —localizado en el cromosoma 8— ni cualquier otro gen sean por sí solos los causantes del envejecimiento.
Los estudios in vitro e in vivo indican que el gen WRN está implicado en la replicación y en la reparación del ADN, así como el mantenimiento de los telómeros3, que son regiones de ADN no codificante, altamente repetitivas, cuya función principal es la estabilidad estructural de los cromosomas en las células eucariotas3 y desempeñan un papel fundamental en la progeria al considerarse un reloj mitótico que regula el número de divisiones celulares4.
En los últimos tiempos ha existido un interés por el estudio de los síndromes de progeria, ya que poseen varias manifestaciones asociadas al envejecimiento, lo que podría dar a conocer una mejor comprensión de los mecanismos moleculares y biológicos implicados en este.
Clínicamente, el SW se caracteriza por el surgimiento del envejecimiento prematuro, asociado a un fenotipo variable, con manifestaciones en múltiples órganos. Durante la infancia y la adolescencia el diagnóstico es poco usual y la característica es el retardo o cese precoz del crecimiento, seguida de una serie de alteraciones que aparecen de forma secuencial.
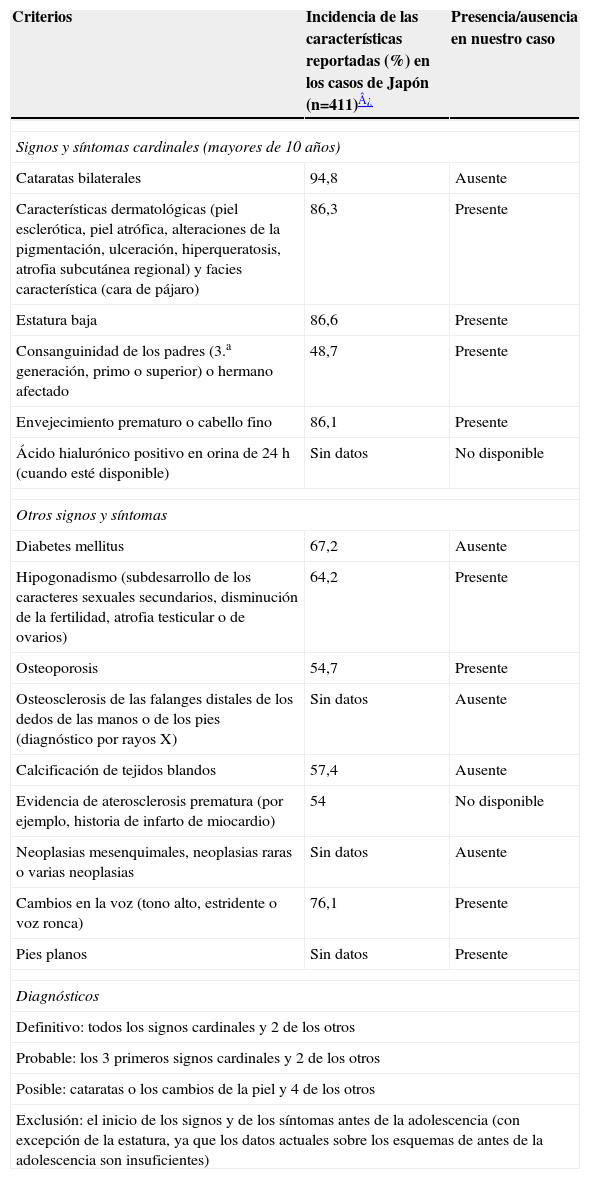
Los criterios diagnósticos del SW se propusieron en 1994 (Nakura et al, 1994)5,6 a partir de un registro internacional de pacientes con SW, y establecen las siguientes posibilidades de diagnóstico: definitivo, probable o posible. El diagnóstico clínico se basa actualmente en la presencia de 4 signos cardinales (cataratas, cambio de la piel, estatura baja y cabello gris o pérdida de este) y la presencia de signos adicionales. En la tabla 1 se describen los criterios diagnósticos tomados del International Registry of Werner Syndrome7.
Criterios diagnósticos (International Registry of Werner Syndrome)
| Criterios | Incidencia de las características reportadas (%) en los casos de Japón (n=411)¿ | Presencia/ausencia en nuestro caso |
| Signos y síntomas cardinales (mayores de 10 años) | ||
| Cataratas bilaterales | 94,8 | Ausente |
| Características dermatológicas (piel esclerótica, piel atrófica, alteraciones de la pigmentación, ulceración, hiperqueratosis, atrofia subcutánea regional) y facies característica (cara de pájaro) | 86,3 | Presente |
| Estatura baja | 86,6 | Presente |
| Consanguinidad de los padres (3.a generación, primo o superior) o hermano afectado | 48,7 | Presente |
| Envejecimiento prematuro o cabello fino | 86,1 | Presente |
| Ácido hialurónico positivo en orina de 24h (cuando esté disponible) | Sin datos | No disponible |
| Otros signos y síntomas | ||
| Diabetes mellitus | 67,2 | Ausente |
| Hipogonadismo (subdesarrollo de los caracteres sexuales secundarios, disminución de la fertilidad, atrofia testicular o de ovarios) | 64,2 | Presente |
| Osteoporosis | 54,7 | Presente |
| Osteosclerosis de las falanges distales de los dedos de las manos o de los pies (diagnóstico por rayos X) | Sin datos | Ausente |
| Calcificación de tejidos blandos | 57,4 | Ausente |
| Evidencia de aterosclerosis prematura (por ejemplo, historia de infarto de miocardio) | 54 | No disponible |
| Neoplasias mesenquimales, neoplasias raras o varias neoplasias | Sin datos | Ausente |
| Cambios en la voz (tono alto, estridente o voz ronca) | 76,1 | Presente |
| Pies planos | Sin datos | Presente |
| Diagnósticos | ||
| Definitivo: todos los signos cardinales y 2 de los otros | ||
| Probable: los 3 primeros signos cardinales y 2 de los otros | ||
| Posible: cataratas o los cambios de la piel y 4 de los otros | ||
| Exclusión: el inicio de los signos y de los síntomas antes de la adolescencia (con excepción de la estatura, ya que los datos actuales sobre los esquemas de antes de la adolescencia son insuficientes) | ||
Al aplicar los criterios diagnósticos, se establece como un caso posible de SW por presentar los cambios en la piel y 4 de los otros signos. Pero es importante reconocer que en la paciente el diagnóstico se establece de una forma precoz y aún no se manifiestan las alteraciones asociadas a este síndrome que hacen parte de los criterios diagnósticos, debido a que estas aparecen de forma secuencial. En la mayor proporción de los casos el diagnóstico suele realizarse a los 36 años de edad, pero este diagnóstico tardío sucede por desconocimiento de la enfermedad. Los estudios moleculares realizados no revelaron mutación del gen WRN. Hoy sabemos que el 10% de estos pacientes que presentan características clínicas no muestra alteraciones en este gen y se consideran como afectados del SWA.
El SWA es otra forma de síndrome progeroide, descrito por primera vez por Lishan Chen en el año 2003, en una paciente de 15 años de edad que presentaba lipodistrofia, hígado graso, diabetes mellitus tipo 2 y características progeroides.
Un informe reciente reveló que el 15% de estos pacientes atípicos tiene mutaciones del gen LMNA, cambio que resulta en sustituciones de aminoácidos en el dominio Lamin1–8. En nuestra paciente tampoco se detectó esta mutación.
En conclusión, creemos que esta paciente muestra una constelación de síntomas más cercano al SW. Aunque el SW no se puede descartar definitivamente, puede ser que esta paciente represente un síndrome progeroide atípico o un síndrome único.
El diagnóstico de SW, a pesar de ser raro, debe acordarse en el diagnóstico diferencial de esclerodermia sistémica, principalmente ante la presencia de manifestaciones atípicas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al International Registry of Werner Syndrome.
