




Las inmunodeficiencias primarias (IDP) son entidades calificadas como «enfermedades raras». Aunque de forma individual su prevalencia es baja, cada año se descubren nuevos genes que asocian inmunodeficiencia. Actualmente hay 340 defectos identificados, cuando hasta el año 2000 se conocían menos de 1001. Posiblemente sean entidades más frecuentes de lo estimado. Se calcula que una de cada 2.000 personas presenta una IDP1,2, prevalencia superior a la de la leucemia en Estados Unidos2.
Las IDP pueden empezar con infecciones recurrentes en la infancia, aunque en ocasiones lo hacen como autoinflamación o autoinmunidad1,3. Estas presentaciones menos esperadas pueden ocasionar retraso diagnóstico, pero su identificación precoz condiciona pronóstico, evitan morbilidad y disminuyen la mortalidad en IDP que precisan trasplante de progenitores3.
En 2012 se creó una consulta especializada en nuestro servicio para el seguimiento de pacientes con IDP confirmada y para atender despistajes en casos sospechosos. Desde entonces hasta 2017 se ha atendido a 135 niños, el 57% de ellos derivados en los últimos 2 años y el 41% en 2017.
Un 57% de los casos tiene una IDP confirmada por nuestro Servicio de Inmunología (77/135), el 15% continúan en estudio (21/135) y en el 27% se ha descartado IDP (37/135). Un 78% de los niños derivados de otros hospitales tenían IDP (25/32), el 50% de los derivados desde otro servicio (39/79) y el 42% desde atención primaria (8/19) (p=0,009). Un 51% fueron derivados por infecciones recurrentes/graves (69/135), un 22% por IDP diagnosticada (30/135), un 19% por linfopenia o hipogammaglobulinemia (26/135), un 6% por antecedentes familiares de IDP (8/135) y el 1,4% por retraso en la caída del cordón (2/135). Un 20% estaban recibiendo previamente a su derivación profilaxis antimicrobiana o gammaglobulina sustitutiva (28/135).
Las IDP diagnosticadas se detallan en la tabla 1 y las características de los pacientes en la figura 1. Un 9% de los pacientes presentaban consanguinidad (6/71). Todos los niños que desarrollaron infección sintomática por citomegalovirus (n=6) presentaron una IDP.
Diagnósticos de los pacientes con IDP en nuestra consulta, según la última clasificación de la International Union of Immunological Societies
| IDP diagnosticadas | Pacientes |
|---|---|
| 1. ID combinadas | 50% (39/77) |
| IDCS | |
| Déficit de ADA | 4 |
| Deficiencia de cadena γ común | 4 |
| Deficiencia de RAG2 | 2 |
| Déficit de CD3δ | 2 |
| Deficiencia de DCLRE1C (Artemis) | 1 |
| Deficiencia de RAG1 | 1 |
| IDCS sin confirmación genética | 2 |
| IDC no severas | |
| Déficit de MHC de clase II | 4 |
| Defecto de CD40 ligando | 1 |
| IDC con características sindrómicas asociadas | |
| Ataxia-telangiectasia | 4 |
| Síndrome de DiGeorge | 4 |
| Síndrome de Kabuki | 2 |
| Disqueratosis congénita | 2 |
| Síndrome de Wiskott-Aldrich | 2 |
| Hipoplasia cartílago-pelo | 1 |
| ID con múltiples atresias intestinales (TTC7A) | 1 |
| Defectos del canal de calcio (ORAI1) | 1 |
| Síndrome hiper-IgE autosómico dominante (STAT3) | 1 |
| 2. Defectos predominantemente de anticuerpos | 34% (26/77) |
| Agammaglobulinemia ligada al cromosoma X | 8 |
| Deficiencia selectiva de IgA | 8 |
| ID variable común | 7 |
| Ausencia de respuesta específica de anticuerpos | 2 |
| Déficit de subclases | 1 |
| 3. Enfermedades por disregulación inmune | 4% (3/77) |
| ALPS-caspasa 10 | 2 |
| Haploinsuficiencia de CTLA-4 | 1 |
| 4. Defectos congénitos del número o función fagocítica | 6% (5/77) |
| Enfermedad granulomatosa crónica | 5 |
| 5. Defectos de la inmunidad innata | 1% (1/77) |
| Deficiencia del receptor β de IL12 | 1 |
| 6. Déficit del complemento | 4% (3/77) |
| Deficiencia de CD46 | 3 |
IDC: inmunodeficiencia combinada; IDCS: inmunodeficiencia combinada severa.
Fuente: Basada en Picard et al.1.
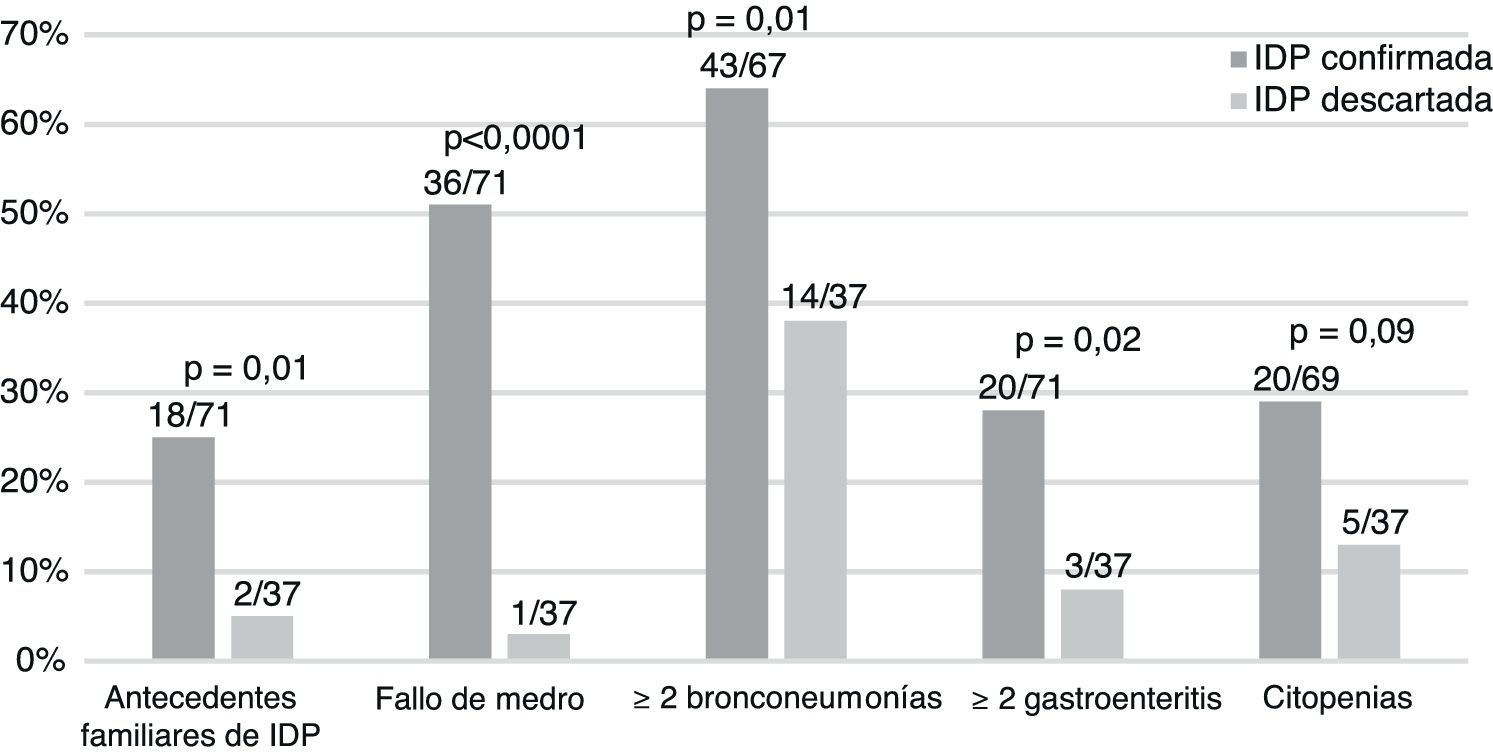
El 34% de los pacientes con IDP precisaron hospitalización en la primera consulta (26/75) vs. el 11% de los finalmente sanos (4/37) (p=0,006). Un 68% (17/25) de los niños con IDP que provenían de otro hospital fueron hospitalizados en su primera consulta, el 18% (7/39) de los derivados desde otro servicio y el 12% (1/8) de los referidos desde primaria (p=0,0001).
Un 57% de los niños con IDP han precisado gammaglobulina (42/74), un 41% profilaxis antimicrobiana (31/75) y el 36% recibieron o precisan trasplante de progenitores (28/77). La mortalidad ha sido del 13% (10/77).
Cada día que pasa se conocen más defectos genéticos que condicionan IDP. Estas conllevan elevada morbimortalidad, especialmente en casos de retraso diagnóstico. Las infecciones recurrentes son motivo frecuente de consulta y establecer qué paciente precisa estudio inmunológico es complejo. El antecedente de consanguinidad es un factor que debe hacer pensar en una posible IDP. Una serie reciente que analiza niños con historia de infecciones recurrentes evidenció que un 21% de ellos presentaban IDP4, elevado porcentaje explicado por una elevada tasa de consanguinidad (38%).
Sin embargo, las infecciones no son siempre la presentación inicial. En los últimos años se han cuestionado los signos clásicos para identificar las IDP5, dado que pueden no identificar pacientes con IDP por desregulación inmune, que empezaban con autoinmunidad o inflamación. Fischer et al. describen como los niños con IDP presentan un riesgo 830, 80 y 40 veces superior de anemia hemolítica, enfermedad inflamatoria intestinal y artritis reumatoide respecto a la población infantil sana6.
Nuestros datos señalan que la presencia de fallo de medro, infecciones sintomáticas por citomegalovirus y antecedentes familiares de IDP son signos de alarma relevantes3-5. De la misma manera, aquellos pacientes más graves, derivados de centros hospitalarios o que requieren ingreso, son pacientes de riesgo.
Recientemente hemos observado un crecimiento de derivación de pacientes, muchos con sospechas diagnósticas consistentes, y más de la mitad con diagnóstico final de IDP. Este incremento es debido al aumento de los pacientes diagnosticados desde nuestro Servicio de Inmunología y al conocimiento progresivo de los médicos derivadores de la existencia de esta consulta especializada.
Los pacientes derivados son vistos en una consulta de Pediatría sin lista de espera. Cuando la derivación es por infecciones recurrentes o sospecha de IDP, son atendidos por pediatras especialistas y por un equipo de inmunólogos clínicos.
Nuestros datos tienen limitaciones. Su carácter retrospectivo hace que algunos datos sean incompletos, por no encontrarse recogidos en algunas historias. Al no ser multicéntrico, ofrece la perspectiva particular de los pacientes atendidos en un centro con unas características concretas. Aun así, nuestros datos muestran la necesidad creciente de este tipo de consulta como recurso para los pediatras, con el fin de proporcionar atención temprana y adecuada a estos niños y disminuir sus morbilidades.
